Реферат: Синтез и исследование сорбционных свойств гуанидинсодержащих полимерных нанокомпозитов
Магистерская диссертация
«Синтез и исследование сорбционных свойств гуанидинсодержащих полимерных нанокомпозитов»
ВВЕДЕНИЕ
Современный этап химической науки характеризуется пристальным вниманием к развитию новых методов получения наноматериалов. Ионообменные сорбенты, модели биополимеров, нанотрубки с повышенной прочностью, органические наноматериалы, обладающие многими свойствами недоступными неорганическим веществам; полимерные нанокомпозитные и пленочные материалы для нелинейных оптических и магнитных систем, газовых сенсоров, биосенсоров, мультислойных композитных мембран; полимерные наноструктуры для гибких экранов – таков далеко не полный перечень практического применения нанокомпозиционных материалов. Можно полагать, однако, что сегодня область разработки и исследования полимерных наноматериалов находится лишь на начальном участке того экспоненциального пути, который предсказывает ей логика развития науки.
В последние годы проводятся широкие исследования процессов получения композиционных материалов на основе полимеров и природных слоистых силикатов. Этот класс новейших нанокомпозитов обладает синергизмом свойств исходных компонентов. Необычность их архитектурного оформления заключается в том, что органическая фаза может захватывать наночастицы внутрь своеобразной «ловушки» или полимерной сетки.
В то же время многие природные силикаты (так называемые смектиты), к числу которых относятся, например, гекторит и монтмориллонит (со структурой типа слюды), состоят из чередующихся слоев катионов и отрицательно заряженных слоев силикатов. Такие слои «хозяина» с регулируемыми системами перколяционных пор и каналов легко образуют соединения включения, в том числе и в случае молекул мономера в качестве «гостей» [1-10].
Одним из наиболее эффективных способов получения нанокомпозитов является внедрение в глинистый минерал мономеров с их постинтеркаляционной полимеризацией [5]. В этом случае образуются нанокомпозиты, в которых неорганический и полимерный компоненты связаны сильными ковалентными или ионными химическими связями.
Перспективными структурами для получения полимерных нанокомпозитов являются полигуанидины. Гуанидинсодержащие полимеры содержат большое количество функциональных групп, которые способны образовывать прочные связи с поверхностью глины. В связи с этим, распределение органоглины в полимерной матрице будет осуществляться гораздо легче, что позволит решить основную проблему, которую приходится преодолевать при создании таких материалов — несовместимость неорганических и органических компонентов. Кроме того, соединения, содержащие гуанидиновые группы, проявляют заметную биоцидность. Введение таких групп в макромолекулярные цепи приводит к усилению биоцидных свойств, поэтому создание новых полимерных композиционных материалов, содержащих гуанидиновые группы, изучение их физико-химических свойств и характеристик, а также выявление возможности получения на их основе нанокомпозитов, обладающих сорбционными и биоцидными свойствами, представляется нам весьма актуальным.
Цели настоящей работы заключались в исследовании возможности получения новых гуанидинсодержащих полимерных композиционных материалов на основе гуанидинсодержащих мономеров методом постинтеркаляционной радикальной полимеризации, и изучении их физико-химических, сорбционных и биоцидных свойств.
ГЛАВА 1. ОБЗОР ЛИТЕРАТУРЫ
Полимер — силикатные нанокомпозиты: структура и свойства
Расширение сфер применения наноматериалов, потребность в них применительно к самым разнообразным отраслям, естественно стимулирует интерес исследователей к проблемам синтеза и механизма образования этого класса полимерных материалов.
К настоящему времени получено достаточно много классов полимерных нанокомпозитов, имеющих различные механизмы упрочнения, но общие в том смысле, что эти механизмы реализуются за счет введения в полимерную матрицу частиц нанометровых размеров. Как известно, основной особенностью таких частиц является резко увеличенная площадь контакта полимер-наполнитель по сравнению с обычными наполнителями размера микронного масштаба, что дает максимальный эффект упрочнения при малых содержаниях нанонаполнителя [11]. Отсюда следует, что основным предметом исследования в данном случае являются межфазные явления на границе полимерная матрица-наполнитель. Взаимодействие на межфазной границе приводит к изменению свойств отдельных компонентов системы, в результате чего композиционные материалы приобретают принципиально новые свойства по сравнению с составляющими их компонентами. Степень гетерогенности и соотношение между фазами часто являются основными для проявления эффекта синергизма. Именно сложность нанокомпозитов и обеспечивает их подчас уникальные свойства как материалов, предназначенных для использования в различных сферах.
Поскольку все процессы, проходящие при получении нанокомпозиционных материалов и при их эксплуатации в различных условиях учесть и описать в настоящее время невозможно, исследователи идут по пути обобщения отдельных экспериментальных фактов, постепенно увеличивая их количество в многопараметрической задаче. Развитие методологических возможностей и создание принципов построения полимерных нанокомпозиционных материалов позволяют заложить основы для создания сложных полимерных систем с необходимым комплексом свойств.
1.1 Общая характеристика композиционных материалов
Композиционный материал (КМ) — это система из двух или нескольких фаз, отличающихся химическим составом и структурой [12-14]. Равномерно распределенные жёсткие компоненты — наполнители играют главную роль в изменении свойств полимера — матрицы в КМ. По форме частиц наполнителя КМ разделяют на дисперсные, слоистые и волокнистые [15].
Полимерные нанокомпозиты — это класс композиционных материалов, представляющих собой полимеры, наполненные частицами, имеющими хотя бы один из размеров нанометрового диапазона. В зависимости от того, какой из размеров имеет такой масштаб, различают три типа наполнителей:
— когда во всех трех измерениях частицы имеют размер порядка нанометра, мы имеем дело с нуль-мерными изоразмерными наночастицами (таковыми, в частности, являются сферические силикатные наночастицы, полученные по in situ золь-гелевой технологии [15-17]). К нуль-мерным наночастицам можно отнести также полупроводниковые нанокластеры [18], магнитные кластеры и др.;
— если лишь два размера частиц наполнителя (удлиненные одномерные образования) имеют нанометровый масштаб, а третий размер – существенно больше, речь идет о нанотрубках [19] или так называемых вискерах (усах) [20, 21], которые широко используются как упрочняющие нанонаполнители с исключительными зачастую уникальными свойствами;
— частицы третьего типа наполнителей (двумерные пластинчатые образования) характеризуется только одним размером нанометрового уровня. В этом случае наполнитель представляет собой слои, толщиной от одного до нескольких нанометров и длиной в сотни, и даже и в тысячи нанометров.
Семейство нанокомпозитов, в которых используются наполнители третьего типа, объединено под названием полимерно-слоистые нанокомпозиты. Эти материалы почти всегда получаются путем интеркаляции (внедрения) цепей полимера (или молекул мономера, впоследствии полимеризующегося) в межслоевые пространства кристалла-хозяина (основного кристалла). Существует множество синтетических и природных слоистых минералов, которые можно использовать в качестве наполнителей для полимеров. В таблице 1 показаны некоторые из них. После интеркаляции полимера в межслоевые пространства большинства из представленных в таблице слоистых кристаллов образуются нанокомпозиты с различными практически важными свойствами [8]. Однако для использования в качестве наполнителей при производстве крупнотоннажных материалов наиболее перспективными среди них оказались природные слоистые алюмосиликаты, прежде всего благодаря таким своим качествам, как доступность, дешевизна, возможность относительно простого регулирования поверхностных свойств [22-24].
Таблица 1.1
Примеры слоистых кристаллических структур, в которых
возможна интеркаляция макромолекул полимера [8]
Химическая природа | Примеры |
Элемент | Графит |
Халькогенидные металлы | (PbS)1,18 (TiS2 ), MoS2 |
Окись углерода | Окись графита |
Фосфаты металлов | Zr(HPO4 ) |
Глины и слоистые алюмосиликаты | Монтмориллонит, гекторит, сапофит, флюоромика, флюогекторит, вермикулит, каолинит, магадит… |
Двойные слоистые гидроксиды | M6 Al2 (OH)16 CO3 ·nH2 O; M=Mg, Zn |
1.2 Структура слоистых алюмосиликатов и их физические и химические свойства
Природные слоистые алюмосиликаты, обычно используемые в нанокомпозитах, принадлежат к структурному семейству, известному, как 2:1-филосиликаты[6, 25, 26].
Атомные структуры обычно встречающихся глинистых минералов были весьма детально определены в результате работ многочисленных исследователей, которые при этом основывались на обобщениях Паулинга [27], сделанных им для структуры слюд и родственных им слоистых минералов.
В основе строения атомных решеток большинства глинистых минералов лежат два структурных элемента. Один структурный элемент состоит из двух слоев плотноупакованных атомов кислорода или гидроксильных групп, между которыми в октаэдрической координации расположены атомы алюминия, железа или магния, так что они равноудалены от 6 кислородов или гидроксилов (рис. 1). Если в этих положениях находятся атомы алюминия, они заполняют лишь две трети всех возможных положений, чтобы уравновесить структуру, которая в этом случае является структурой гидраргиллита и имеет формулу А12 (ОН)6. При наличии магния заполненными оказываются все положения; уравновешенная структура является в этом случае структурой ору сита и характеризуется формулой Mg3 (OH)6. Обычно расстояние О — О равно 2,60 Å, а расстояние ОН — ОН обычно 3 Å, однако в данном структурном элементе расстояние ОН — ОН имеет величину 2,94 Å, и пространство, доступное для иона в октаэдрической координации, имеет размер примерно 0,61 Å. Толщина этого структурного элемента в структурах глинистых минералов равна 5,06 Å. Второй элемент структуры построен из кремнекислородных тетраэдров. В каждом тетраэдре атом кремния равноудален от четырех атомов кислорода или гидроксильных групп в зависимости от требований баланса структуры, образованной тетраэдрами с атомами кремния в их центрах. Тетраэдрические группы кремнезема расположены в форме бесконечно повторяющейся гексагональной сетки, образующей слой состава Si4 О6 (OH)4 (рис. 1). Тетраэдры расположены так, что вершины каждого из них направлены в одну и ту же сторону, а основания находятся в одной и той же плоскости. В структуре этого слоя можно выделить три уровня. На первом уровне в виде ажурной сетки находятся атомы кислорода. На втором уровне находятся атомы кремния, каждый из которых лежит в выемке, образованной тремя соприкасающимися атомами кислорода первого уровня, вследствие чего они в своей совокупности образуют гексагональную сетку. На третьем уровне находятся гидроксильные группы, причем каждый гидроксил расположен в вершине тетраэдра прямо над атомом кремния. Гранецентрированная гексагональная сетка может рассматриваться как образованная тремя системами рядов атомов кислорода, пересекающихся под углом в 120°. Расстояние О — О в тетраэдрическом слое равно 2,55 Å, а пространство, доступное для иона, в тетраэдрической координации имеет размер примерно в 0,55 Å. Толщина этого структурного элемента в структурах глинистых минералов — 4,93 Å; каждый из них имеет расстояние от центра до центра примерно 2,1 Å [28].
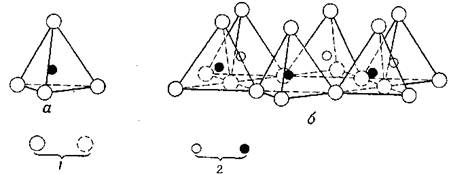
Рис. 1.1. Схематическое изображение отдельного кремнекислородного тетраэдра (а) и сетки кремнекислородных тетраэдров, расположенных по гексагональному мотиву (б): 1 – атомы кислорода; 2 – атомы кремния
Некоторые глинистые минералы являются волокнистыми и построены из структурных элементов, отличных от описанных выше. По своим структурным особенностям эти минералы подобны амфиболам, и их основные структурные элементы состоят из кремниевых тетраэдров, расположенных в форме двойной цепи состава Si4 О11, как показано на рис. 2. Структура этого элемента подобна структуре слоя из кремнекислородных тетраэдров в слоистых минералах с тем лишь отличием, что она имеет бесконечную протяженность только в одном направлении. В другом направлении она распространена лишь примерно на 11,5 Å. Цепи связаны вместе атомами алюминия или магния, расположенными так, что каждый из них окружен шестью «активными» атомами кислорода. Активными являются те атомы кислорода, которые имеют лишь одну связь с кремнием и, следовательно, находятся на краях цепей и в вершинах тетраэдров [29].
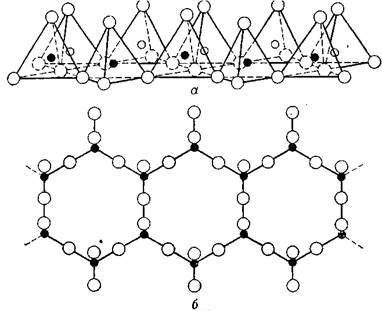
Рис. 1.2. Схематическое сдвоенных цепей кремнекислородных тетраэдров, характерных для амфиболевого структурного типа глинистых минералов. а – в перспективе;
б – в проекции на плоскость основания тетраэдров
1.2.1 Монтмориллонитовые минералы
Монтмориллонитовые минералы встречаются лишь в виде частиц крайне малого размера, так что они не могут быть исследованы рентгеновскими методами, пригодными для изучения монокристаллов. Концепции об их структуре, таким образом, должны основываться на данных порошковых рентгенограмм и учете особенностей более детально изученных структур. В связи с этим имеется неопределенность в отношении структуры монтмориллонита.
Представления о характере структуры монтмориллонитовых минералов впервые высказали в 1933 г. Гофманом, Энделл и Вилмом. В дальнейшем они были несколько изменены, согласно гипотезам Мегдсфрау и Гофмана [27], Маршалла [30] и Хендрикса [28]. Согласно этим представлениям, монтмориллонит состоит из структурных элементов, построенных из двух наружных кремнекислородных тетраэдрических сеток и промежуточной алюмокислородной октаэдрической сетки. Все вершины тетраэдров в сетке направлены в одну и ту же сторону — к средней части структурного элемента. Тетраэдрические и октаэдрические сетки сочленены друг с другом таким образом, что вершины тетраэдров каждой кремнекислородной сетки совместно с вершинами слоев гидроксилов октаэдрической сетки образуют общий слой. В вершинах, общих для тетраэдрических и октаэдрических. сеток, располагаются вместо гидроксильных групп ОН атомы О. Слои являются непрерывными в направлениях а и b и наложены друг на друга в направлении с.
Во взаимном расположении кремне- и алюмокремнекислородных слоев слои атомов кислорода каждого структурного элемента являются смежными со слоями атомов кислорода соседних структурных элементов, вследствие чего между ними существует очень слабая связь и в минерале наблюдается прекрасная спайность. Характерной особенностью структуры монтмориллонита является то, что молекулы воды и другие полярные молекулы, например некоторые органические молекулы, могут проникать между структурными слоями, вызывая расширение решетки в направлении оси с. В связи с этим величина периода с не является постоянной, а изменяется, начиная примерно с 9,6 Å, когда между структурными слоями нет никаких полярных молекул, до полного, в некоторых случаях материального разобщения отдельных слоев. На рис. 3. схематически изображена структура монтмориллонита такого типа. Обменные катионы находятся между силикатными слоями, и межплоскостное расстояние вдоль оси с полностью дегидратированного монтмориллонита в некоторой степени зависит от размера межслоеных катионов: оно тем больше, чем больше по своим размерам катионы. В случае адсорбции полярных органических молекул между силикатными слоями величина периода с также изменяется и зависимости от размера и геометрии органической молекулы. Толщина водных слоев, расположенных между силикатными структурными слоями, при данном давлении паров воды зависит от природы обменных катионов.
В нормальных условиях монтмориллонит с Na+ в качестве обменного нона обычно имеет между силикатными слоями один слой молекул воды; межплоскостное расстояние в направлении оси с примерно равно 12,5 Å. Монтмориллонит с Са++ обычно содержит два молекулярных слоя воды и имеет межплоскостное расстояние в направлении оси с 15,5 А. Свойства разбухания монтмориллонитов являются обратимыми. Однако если структура полностью сжата при удалении всех межслоевых полярных молекул, повторное набухание может происходить с трудом.
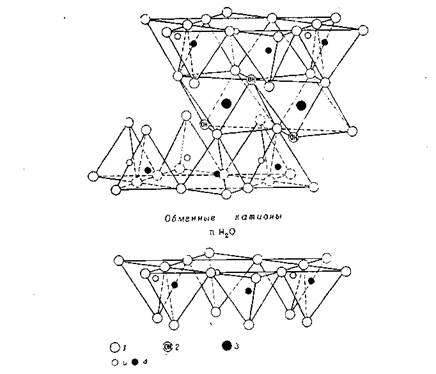
Рис. 1.3. Схематическое изображение структуры монтмориллонита (по Гофману, Энделлу, Вилму, Маршаллу и Хендриксу)
Опыты Меринга [31] и других авторов [32] с монтмориллонитом (в присутствии большого количества воды) указывают, что при одних поглощенных катионах, например Nа+, структурные слои могут полностью разойтись, а при других катионах, таких, как Са++ и Н+, разделение слоев не является полным.
Как впервые было отмечено Маршаллом [30] и Хендриксом [28], состав монтмориллонита всегда отличается от состава, выражаемого вышеприведенной теоретической формулой, вследствие замещения в пределах решетки кремния в тетраэдрической координации алюминием и, возможно, фосфором или алюминия в октаэдрической сетке магнием, железом, цинком, никелем, литием и т. д. В тетраэдрической сетке замещение Si4+ на А13 + ограничено, по-видимому, 15%. Согласно приведенной выше формуле, в монтмориллоните заполнены лишь две трети возможных положений в октаэдрической сетке. Замещение А13+ на Mg2+ может быть в соотношении один к одному или 2А13+ на 3Mg+2, причем в последнем случае заполняются все октаэдрические положения. В октаэдрической сетке степень замещения изменяется от самых малых значений вплоть до полного замещения. Полное замещение 2А13+ на 3Mg2+ дает минерал сапонит; замещение алюминия железом — нонтронит; хромом — волконскоит, цинком — соконит.
Радиус иона Mg++ равен 0,65Å, а иона Fe3+ — 0,67Å. Эти ионы слишком велики, чтобы строго соответствовать решетке монтмориллонита; в результате монтмориллонитовые минералы со значительными замещениями этими ионами подвержены направленным напряжениям, проявляющимся в удлиненной щепкоподобной или игольчатой форме частиц.
Слоистые минералы, у которых все возможные октаэдрические положения заполнены, в общем случае называются октафиллитами, или триоктаэдрическими, тогда как минералы, у которых заполнены лишь две трети возможных октаэдрических положений, называются гептафиллитами, или диоктаэдрическими. Многочисленные анализы монтмориллонита доказали, что благодаря замещениям в пределах октаэдрического слоя этот минерал является либо триоктаэдрическим, либо диоктаэдричсским. Росс и Хендрикс [27] произвели пересчеты большого числа химических — анализов монтмориллонита для приведения их в соответствие с его структурой. К сожалению, эти пересчеты не сопровождаются структурными данными и некоторые из образцов могут оказаться смесью глинистых минералов. Однако выводы, сделанные на основании этих анализов, можно считать правильными, так как количество анализов достаточно велико. Согласно Россу и Хендриксу, число ионов в шестерной координации, т. е. ионов октаэдрических положений, находится в двух пределах: от 4.00 до 4,44 и от 5.76 до 6,00. Далее, если минерал является диоктаэдрическим, то для него, по-видимому, возможна значительная вариация точного расположения алюминия и других атомов ни всем возможным октаэдрическим положениям.
Другое обстоятельство, в силу которого монтмориллонит всегда отличается от идеальной формы, выражаемой его теоретической формулой, заключается в том, что его решетка вследствие указанных выше замещений Аl3+ на Mg++, Si4+ на Аl3+ и т. д. всегда неуравновешенна. Такая неуравновешенность может обусловливаться замещениями ионов разной валентности как в тетраэдрической, так и в октаэдрической сетках. Неуравновешенность в одной из этих сеток может компенсироваться частично (но лишь частично) замещениями в других сетках структурного слоя. Так, замещения Si4+ на Аl3+ могут быть частично компенсированы за счет заполнения более двух третей октаэдрических положений. Компенсация может также происходить благодаря замещениям атомов кислорода октаэдрического слоя на группы ОН. Существенно, что замещения в решетке монтмориллонита совместно с внутренними компенсирующими замещениями всегда вызывают почти один и тот же результирующий отрицательный заряд решетки. Согласно данным многих анализов, такой заряд равен примерно 0,66 на элементарную ячейку. Результирующий отрицательный заряд уравновешивается обменными катионами, адсорбированными между структурными слоями и вокруг их краев (ионный обмен), и равен примерно двум третям единицы на элементарную ячейку. Для его достижения требуется, например, замещение каждого шестого А13+ на Mg2+ или каждого шестого Si4+ на А13+ .
Отрицательный заряд монтмориллонита, синтезированного из чистых водных смесей окиси магния и кремнезема, не мог быть вызван замещениями в решетке. Он мог быть обусловлен наличием вакантных мест в решетке; такие вакантные места, возможно, имеются у естественных минералов.
Росс и Хендрикс [27] по данным химического анализа вычислили структурные формулы многих монтмориллонитов, что позволило установить пределы замещений в решетке и характер частично компенсирующих замещений в структуре. Номенклатура минералов монтмориллонитовой группы зависит от характера изоморфных замещений в решетке. В табл. 1.1 приведены, согласно Россу и Хендриксу [27], наименования монтмориллонитов и соответствующие им структурные химические формулы.
Таблица 1.2.
Формулы некоторых членов монтмориллонитовой группы, выведенные Россом и Хендриксом
Диоктаэдрические (гептафиллитовые) монтмориллониты | |
монмориллонит | (ОН)4 Si3 (Al3,34 Mg0,66 {Na0,66 })O20 |
Бейделлит (1) | (ОН)4 (Si4,34 Al1,66 {Na0,66 }) Al4,34 O20 |
Бейделлит (2) | (ОН)4 (Si6 Al2 {Na0,56 }) Al4,44 O20 |
Нонтронит (алюминиевый) | (ОН)4 (Si7,34 Al0,66 {Na0,66 }) Fe4 3+ O20 |
Триоктаэдрические (октафиллитовые) монтмориллониты | |
гекторит | (ОН)4 Si6 Mg5,04 {Na0,66 }Li0,66 )O20 |
сапонит | (ОН)4 (Si7,34 Al0,66 {Na0,66 })Mg6 O20 |
Саполит (алюминиевый) | (ОН)4 (Si6,66 Al1,34 {Na0,66 })(Mg5.34 Al0,66 ) O20 |
Эти наименования полностью соответствуют их обычному употреблению, за исключением бейделлита, существование которого в качестве самостоятельного минерала многими исследователями подвергается сомнению. Фигурные скобки в структурных формулах помещены с той группой, которая имеет отрицательный заряд, требующий для уравновешения структуры добавления катиона, внешнего по отношению к силикатному слою.
Мак-Эван [33] проанализировал связь размеров а и b элементарной ячейки монтмориллонита с изменениями его химического состава. Он сделал заключение о том, что длина осевых периодов должна возрастать в порядке монтмориллонит ® нонтронит ® сапонит. Он предложил следующую формулу для вычисления b0, из которой также может быть получено значение а0:
b0= 8,91 + 0,06r + 0,34s + 0,048t Å
где r — число ионов А1 в тетраэдр и чес ко и координации;
s — число ионов Mg в октаэдрической координации;
t — число ионов Fe в октаэдрической координации (в каждом случае указаны числа на элементарную ячейку).
Эта формула основана на значениях, полученных для мусковита, талька и нонтронита; предполагается, что изменения периодов, связанные с указанными выше замещениями, пропорциональны и аддитивны. Согласно данным Мак-Эвана [33], формула достаточно хорошо согласуется с экспериментальными значениями; изменения b0в значительно большей степени зависят от характера заселения октаэдрических положений, чем от заселении тетраэдрических положений.
Отношение молекулярных количеств кремнезема и глинозема и решетке монтмориллонита может изменяться в пределах примерно от 1:1 до 1:3. В первом случае имеет место максимальное заполнение октаэдрических положений ионами Аl3+ (около 4,44 на элементарную ячейку) с дополнительным замещением Si4+ на А13+. Замещение Si4 + на А13+ вызывает неуравновешенный заряд, который компенсируется избыточным зарядом октаэдрической сетки. В случае наиболее высокого отношения псе тетраэдрические положения заселены ионами Si4+, минерал является диоктаэдрическим с максимальной степенью замещения Al3+ каким-либо двухвалентным ионом для обеспечения отрицательного заряда слоев.
Железо может, по-видимому, замещать алюминий во всех его положениях в октаэдрической сетке и совершенно не замещает его в тетраэдрической сетке. Богатые железом разновидности монтмориллонита — нонтрониты, насколько можно судить по имеющимся анализам, обнаруживают незначительную степень замещения ионов Fe3+ ионами Mg2+, так что отрицательный заряд слоев вызван главным образом замещениями ионов Si4+ ионами А13+. В триоктаэдрических монтмориллонитах отрицательный заряд обусловлен в основном замещениями Si4+ на А13+. Можно считать на основании данных Росса и Хендрикса [34], что у триоктаэдрических монтмориллонитов в октаэдрической сетке присутствует примерно до одного атома алюминия или железа па элементарную ячейку. Избыточный положительный заряд октаэдрической сетки уравновешивается отрицательным зарядом тетраэдрической сетки, обусловленным повышенным замещением Si4 + на Аl3+ .
Неоднократно высказывались соображения о том, что структура монтмориллонита, по Гофману и др. [31], Маршаллу [30] и Хендриксу [28], не совсем отвечает всем свойствам этого минерала, в особенности его емкости ионного обмена. Эдельман и Фавейе [35] предложили для монтмориллонита другую структуру, которая якобы объясняет эти свойства более удовлетворительно. Эта структура отличается от структуры, предложенной Гофманном и др. [27], тем, что каждый второй кремнекислородный тетраэдр обеих кремнекислородных сеток перевернут, так что половина тетраэдров направлена в противоположную сторону. Те из них, которые обращены наружу от силикатного слоя, должны иметь в своих вершинах вместо кислорода группы ОН. В этой структуре атомы кремния расположены не в единой плоскости кремнекислородной тетраэдрической сетки; кроме того, некоторые атомы кислорода октаэдрической сетки для сохранения баланса структуры должны заместиться на гидроксильных группах (рис. 4 и 5). Слой имеет следующее распределение зарядов по уровням:
2(OH) — 2-
2Si4+ 8+
6O 12-
2Si4+ 8+
2O 4(OH) 8-
4Al3+ 12+
2O 4(OH) 8-
2Si4+ 8+
6O 12-
2Si4+ 8+
2(OH) — 2-
Межслоевые Н2 О или другие полярные группы

Рис. 1.4. Схематическое изображение структуры монтмориллонита
(по Эдельману и Фавейе)
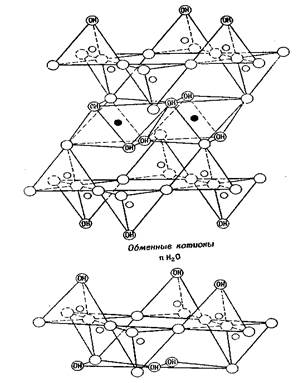
Рис. 1.5. Схематическое перспективное изображение структуры монтмориллонита (по Эдельману и Фавейе)
Этому соответствует структурная формула (OH)12 Si8 Al4 016 • nН2 0 (межслоевая вода). Для этой структуры нет необходимости предполагать какие-либо замещения в пределах решетки для объяснения обменной способности. Решетка может быть полностью уравновешена. Как полагают авторы, обменная реакция связана, прежде всего, с замещением атомов Н из групп ОН, расположенных в наружных вершинах тетраэдров. Согласно первоначальной модели монтмориллонита число таких групп ОН значительно превышало количество их, требуемое значением емкости обмена. Эдельман и Фавейе [36] вынуждены были предположить, что лишь часть их доступна для обмена. Как следует из рентгеновских данных, основанных на синтезах Фурье [36], данных химических анализов, указывающих на наличие изоморфных замещений, и тщательных исследований дегидратации монтмориллонита, структура этого минерала, по Эдельман у и Фавейе, в своем первоначальном варианте [37] не соответствует действительности. Впоследствии Эдельман [38] предложил видоизмененную структурную схему монтмориллонита, согласно которой лишь 20% тетраэдров являются обращенными, что ликвидирует расхождение с экспериментальными значениями емкости обмена. Весьма сомнительно, смогут ли рентгеновские данные подтвердить справедливость этой видоизмененной структурной модели. Такая структура монтмориллонита не совсем согласуется с химическими данными, указывающими на наличие изоморфных замещений, и с геометрией поглощения органических молекул. Характер метиляции отдельных органических веществ при их поглощении монтмориллонитом, указывающий на большее число групп (ОН), чем это совместимо со структурой монтмориллонита, по Гофману и др. [27], заставил некоторых исследователей (Бергер [35], Дуэлл [36]) отдать предпочтение этой структуре. Необходимы Дальнейшие исследования для выяснения возможных структурных изменений, связанных с такими органомонтмориллонитовыми реакциями.
Мак-Коннелл [33] несколько видоизменил структуру монтмориллонита, предложенную Гофманном и др. [27]. Он предположил, что некоторые кремнекислородные тетраэдры структуры замещены тетраэдрами (ОН)4 — что равносильно предположению о наличии пробелов в размещении атомов кремния по тетраэдрическим положениям — с соответствующей заменой атомов О группами ОН для сохранения баланса в структуре. Это должно обеспечить избыточное количество поверхностных групп ОН, необходимое для объяснения некоторых особенностей поглощения органических веществ монтмориллонитом, а также согласуется с данными дегидратации монтмориллонита. Необходимы дальнейшие исследования для подтверждения предположений Мак-Коннелла.
1.3 Органомодификация монтмориллонита
Особый интерес для получения нанокомпозитов представляют минералы, способные к разбуханию (смектиты) [6, 25, 26]. Один из представителей смектитов — монтмориллонит, характерной особенностью которого является способность набухать в некоторых растворителях и диспергироваться на отдельные нанослои при определенной обработке.
Катионы металлов, находящиеся в природном минерале, могут заменяться на другие ионы при проведении реакции ионного обмена. По способности к замещению они могут быть расположены в следующий ряд [39-41]:
А1> Са>К> [(C4 H9 )4 N]> [(C2 H5 )4 N]> [(СНз)4 N]>NH4 >Na> Li
Как отсюда следует, четвертичные алкиламмониевые катионы могут вытеснять ионы Na+ с обменных позиций в монтмориллоните, причем увеличение числа углеродных атомов в неполярных алифатических группах способствует более эффективному вытеснению межслоевых катионов [41]. В связи с этим, чаще всего в качестве модификаторов поверхностных свойств глины используют катионные поверхностно-активные вещества (ПАВ), в которых число углеродных атомов составляет от 6 до 20.
Все глинистые минералы обладают определенной емкостью катионного обмена (ЕКО). Эта величина обозначает количество обменных катионов (выраженное в мг-эквивалентах), способных к замещению на катионы другого типа в расчете на 100 г глины. Монтмориллонит обладает самой высокой среди глинистых минералов емкостью катионного обмена (до 150мг.экв/100г).
Способность катионов металлов в межслоевых пространствах замещаться на катионные ПАВ позволяет модифицировать поверхностные свойства силикатных пластин [42]. Для придания гидрофильным глинам органофильности используют ПАВ с длинными алифатическими цепями. Модифицированная в результате хемосорбции глина (или органоглина) являясь органофильной, имеет меньщую поверхностную энергию и лучше совмещается с органическими полимерами. Когда межплоскостные катионы после ионного обмена замещены более объемными алкиламмониевыми органическими катионами, происходит также и увеличение межслоевого расстояния.
Процессы сорбции органических катионов на глинистых минералах, в том числе на монтмориллоните изучаются уже сравнительно давно [42]. Поскольку в слое силиката присутствует отрицательный заряд, катионная концевая группа алкиламмониевого катиона предпочтительней располагается на поверхности слоя, оставляя алифатическую цепь направленной от или вдоль поверхности. Как отмечено в работе [43], обмен ионов сложных органических соединений во многом отличается от обычного обмена ионов металлов, так как наряду с электростатическим взаимодействием поверхности минерала и иона, проявляется действие ван-дер-ваальсовых сил. В этой же работе указывается и на возможность образования слоев органических катионов внутри межпакетных пространств монтмориллонита. Они могут располагаться более чем одним слоем, так как одного слоя объёмных катионов может быть недостаточно для нейтрализации заряда на поверхности пластин. В результате сорбции объемных органических катионов и десорбции малых ионов натрия, происходит увеличение межплоскостного расстояния между пластинами глины [44, 45].
Межплоскостное расстояние в органоглинах также зависит от ЕКО слоистого силиката. Количество обменных позиций на поверхности силикатных пластин определяет плотность упаковки молекул модификатора.
Как видно из рисунка 6, межпакетное расстояние монтмориллонита увеличивается ступенчато в зависимости от количества атомов углерода в цепи модификатора первичного амина.
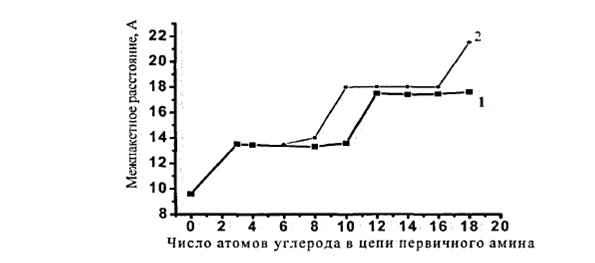
Рис.1.6. Изменение межпакетного расстояния монтмориллонита, модифицированного первичным амином, в зависимости от числа углеродных атомов в цепи амина Ио емкости катионного обмена: 1- малые ЕКО (< 90 мг·экв/100г; 2- большие ЕКО (>90 мг·экв/100г)
Для глин, имеющих различное количество обменных позиций, увеличение межпакетного расстояния при эквивалентной сорбции алифатических аминов с длиной цепи от 1 до 3 метильных групп происходит на величину ~ 0,4 нм (см. рис. 6). Это межплоскостное расстояние примерно равно диаметру алифатических цепей, которые располагаются параллельно пластинам слоистого силиката (рис. 3, а). При дальнейшем увеличении количества метильных групп в алифатических цепях вновь происходит возрастание величины межпакетного расстояния на ~ 0,4 нм при длине углеродной цепи 8-10 атомов для силикатов с малым значением ЕКО и 16-18 для силикатов с большой ЕКО (>90мг*экв/100г). Это может соответствовать переходу алкильных цепей модификатора, находящихся в межслойном пространстве от монослоя к бислою, а затем к образованию псевдотройного слоя. На рисунке 3 показано изменении структуры слоев модификатора [46]. По-видимому, алифатические цепи таких модификаторов способны образовывать бислои, также лежащие параллельно поверхности частицы.
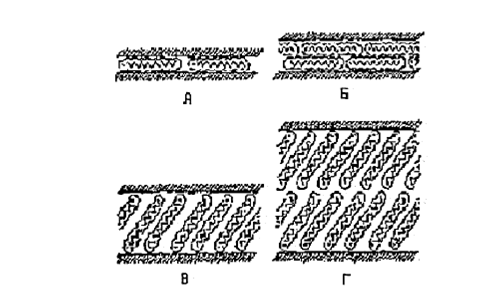
Рис.1.7. Агрегация алкильных цепей в слоистых силикатах: а)горизонтальный монослой; б)горизонтальный бислой; в) монослой «парафинового типа» 3) бислой «парафинового типа»[46]
Авторы работы [48] предполагают, что в случае большой катионной ёмкости, молекулы модификаторов могут образовывать моно- или бислои, которые располагаются в межплоскостном пространстве под определенным углом к поверхности слоистого силиката, т.н. «парафиновый» тип упаковки, а также образовывать гибридные образования, включающие как горизонтальные, так и «парафиновые» фрагменты.
Подробное изучение процессов сорбции различных четвертичных алкиламмониевых катионов на поверхности монтмориллонита было проведено в работах [44, 47]. В работе [48] был проведен анализ структуры слоев модификатора по сдвигу частот валентных асимметрических колебаний СН2 -группы. Оказалось, что внедренные цепи ПАВ существуют в состояниях, характеризующихся различной степенью упорядоченности.
По мере уменьшения плотности упаковки молекул модификатора, уменьшения длины цепи или увеличения температуры внедренные цепи образуют всё более слабо упорядоченную структуру, следствием чего является увеличение соотношения гош/транс конформеров. При определенных значениях доступной поверхности в расчете на одну молекулу оказывается, что упаковка цепей не полностью разупорядочена, а сохраняет некоторый ориентационный порядок аналогично жидкокристаллическому (ЖК) нематическому состоянию (Рис. 8). Следует отметить, что при всей логичности сделанных авторами выводов достоверность экспериментальных данных невелика, так как сдвиги частот, по которым судили об изменении упорядочения молекул, близки к разрешению спектральных приборов.
Методом молекулярно-динамического моделирования было установлено, что по мере удлинения цепи структура прослойки изменяется пошагово от неупорядоченного к более упорядоченному монослою, затем скачкообразно переходя к более беспорядочному псевдодвойному слою (Рис. 1.8).
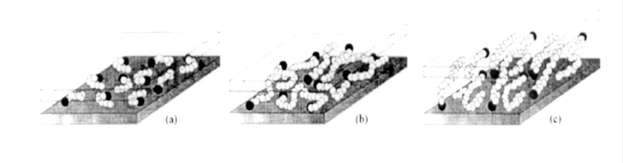
Рис. 1.8. Модели упаковки алкильных цепей: (а) короткие алкильные цепи:
отдельные молекулы, горизонтальный монослой, (б) цепи средней длины:
плоскостная неупорядоченность и образование встречно-штыревой
структуры с формированием «псевдобислоя», (в) длинная цепь: повышенный
межслоевой порядок, жидкокристаллический тип среды [46].
Одной из причин перспективности применения глин в качестве наполнителя является потенциальная возможность перехода их частиц к наноразмерам не за счет механического дробления, а, в основном, посредством химической модификации их поверхности. Кроме этого, для обеспечения высоких физико-механических свойств нанокомпозитов полимер-глина необходима хорошая совместимость органического и неорганического компонентов, которые изначально термодинамически не совместимы.
Для достижения обеих этих целей используется модификация поверхности частиц глины посредством ПАВ. Применение ПАВ должно сформировать между частицами глины органофильные слои, которые снижают поверхностную энергию на границе раздела фаз, увеличивают расстояние между силикатными слоями и, тем самым, облегчают проникновение полимерных цепей в межплоскостные пространства глины.
Подбор ПАВ требует знания особенностей структуры глинистых минералов и индивидуален для каждого полимера.
1.4 Типы полимер-силикатных нанокомпозитов
Правильное использование глин с различной емкостью катионного обмена, подбор модифицирующих органических катионов, направленное формирование органофильных слоев для каждого метода приготовления композитов с учетом индивидуальных свойств полимерной матрицы, позволяет успешно получать нанокомпозиты нескольких типов.
На рисунке 1.9 схематически изображены морфологические образования глины и возможные типы структуры наполнителя, возникающие при взаимодействии слоистых силикатов и полимеров. Для получения нанокомпозитов необходимо разрушить всю иерархию морфологических образований глины вплоть до отдельных кристаллитов и осуществить проникновение полимера в межплоскостное пространство.
Когда полимер не может интеркалировать в пространство между слоями кристаллита, получают композит с разделенными фазами и его основные характеристики лежат в том же диапазоне, что и у обычных микрокомпозитов (рис.4). Кроме этого случая можно выделить два других типа композитов. Первый из них обладает структурой, в которой отдельные вытянутые полимерные цепи интеркалированы в межслоевые пространства глины, формируя тем самым хорошо упорядоченную многослоевую систему, собранную из чередующихся полимерных и силикатных слоев. В композитах второго типа в алюмосиликатные слои, полностью и однородно диспергированны в полимерной матрице, формируют так называемую эксфолиированную или деламинированную структуру [8].
Тип композита на основе полимеров и слоистых алюмосиликатов можно определить, методом рентгеноструктурного анализа (РСА) по угловому положению пика базального рефлекса на рентгенограмме. Сдвиг базального рефлекса в область углов дифракции меньших, чем у исходной глины, подтверждает получение интеркалированного нанокомпозита (ИН), в котором хорошо сохраняется повторяющаяся многослойная структура. Отсутствие дифракционных максимумов — либо из-за большего расстояния между слоями (превышающего 8 нм), либо из-за того, что силикатные пластинки разупорядочены, означает формирование эксфолиированного нанокомпозита (ЭН).
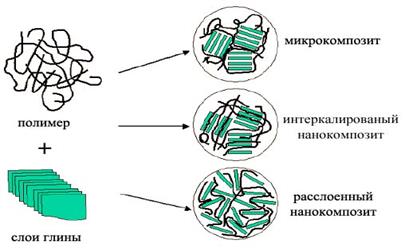
Рис. 1.9. Схематическое изображение морфологических образований глины и типов структуры нанокомпозитов, возникающей при взаимодействии слоистых силикатов и полимеров [48].
Оценить тип композита можно также с помощью просвечивающей микроскопии. Однако результаты, полученные этим методом, достоверны лишь при анализе большой совокупности фотографий.
Следует отметить, что характер упаковки молекул модификатора в межслоевом пространстве определяет величину расстояния между силикатными пластинами, органофильность межслоевых пространств глины, и, в конечном счёте, возможности для получения интеркалированной и эксфолиированной структуры. Однако экспериментальных работ в этой области пока недостаточно [50, 51]. Теоретические исследования, базирующиеся на данных экспериментов, в настоящее время не позволяют достоверно описать структуру адсорбционных слоев в межплоскостных пространствах глины [52-54].
1.5 Методы получения полимер-силикатных нанокомпозитов
Существует несколько методов получения полимер-слоистых силикатных нанокомпозитов [8,55]:
-смешение в растворе. Слоистые силикаты могут набухать в некоторых растворителях. В растворителе слоистый силикат расслаивается на отдельные слои, в котором также растворяется полимер (или форполимер в случае нерастворимого полимера типа полиимида). Затем полимер адсорбируется на раздвинутых силикатных листах, и после испарения растворителя (или осаждения смеси) формируется композит;
— интеркаляционная полимеризация in situ. При получении композитов этим методом слоистый силикат набухает в жидком мономере (или в растворе мономера), таким образом, что образование «полимера может происходить между слоями силиката. Полимеризация инициируется нагреванием или излучением, либо диффузией подходящего инициатора или органического инициатора или предварительно иммобилизованным в межслойном пространстве глины катализатором;
— матричный синтез. Этот метод, в котором силикаты образуются in situ в водном растворе, содержащем полимер и строительные блоки силиката, широко используется для синтеза нанокомпозитов основанных на двухслойных гидроксидах, но гораздо менее развит для слоистых силикатов. В данной методике, рост частиц происходит благодаря самоорганизации отдельных атомов в среде полимера. При этом полимер регулирует рост неорганических кристаллов. Композит формируется в результате того, что полимер застревает между силикатными слоями в процессе их роста;
— смешение в расплаве. Слоистый силикат смешивают с полимерной матрицей в расплаве. При условии, что поверхности слоев совместимы с выбранным полимером, он может проникать в межслойное пространство и образовывать либо ЭН, либо ИН материал. Смешение в расплаве позволяет перерабатывать полимеры, которые нельзя использовать в методах полимеризационного наполнения.
Первую попытку описания процессов интеркаляции макромолекул в межпакетные пространства глины с адсорбированными на силикатных пластинах ПАВ предприняли R. Vaia и Е. Giannelis [56, 57]. Авторы, основываясь на положениях статистической термодинамики и оценивая свободную энергию системы, состоящей из модифицированного слоистого силиката и расплава полимера, получили аналитические выражения для изменения внутренней энергии (вызванного появлением новых межмолекулярных взаимодействий) и энтропии (связанного с изменением конфигурации элементов системы полимер — ПАВ при интеркаляции цепей полимера и раздвижении слоев силиката). Они показали, что изменение энтропии в процессе формирования нанокомпозита происходит в результате двух причин: 1) заключение молекул полимера, раньше нахолившихся в состоянии расплава, в узкий зазор между слоями силиката; 2)изменение конфигурации цепей модификатора. Изменение энтропии модификатораположительно при интеркаляции макромолекул в межплоскостное пространство, т.к. молекулы модификатора приобретают большую конфигурационную свободу по мере увеличения межпакетного пространства, и в значительной степени зависит от поверхностной плотности привитых цепей и их длины. Изменение энтропии полимера, напротив, будет иметь отрицательное значение, поскольку макромолекулы из расплава переходят в узкий зазор и испытывают ограничивающее влияние стенок. Оценка, проведенная авторами модели, показала, что суммарное изменение энтропии системы силикат-модификатор-полимер отрицательно, а значит, формирование нанокомпозита становится неблагоприятным с точки зрения энтропийного фактора. Внутренняя энергия рассматриваемой системы складывается из парных взаимодействий трех компонентов: полимер-силикат, полимер-модификатор и силикат-модификатор. Зная характеристики матричного полимера, можно подобрать тип силиката и модификатора таким образом, чтобы система силикат-модификатор-полимер была максимально совместимой.
Для формирования термодинамически стабильного нанокомпозита необходимо, чтобы энергетические взаимодействия в системе компенсировали уменьшение энтропии. Если энергетические взаимодействия в системе слишком слабы и не позволяют преодолеть энтропийный барьер, будет происходить фазовое разделение компонентов и формироваться обычный микрокомпозит.
Авторы работ [58-60] проводили моделирование взаимодействия в системе силикат-модификатор-расплав полимера на основе теории самосогласованного поля. Взаимодействие между компонентами системы в этом подходе учитывали с помощью параметра взаимодействия Флори-Хаггинса. Вид потенциала взаимодействия определяется плотностью распределения звеньев полимера и модификатора и значениями парных параметров взаимодействия. Анализировали зависимость свободной энергии системы от длины цепи модификатора и полимера, поверхностной плотности молекул модификатора. Результаты, полученные этой группой исследователей, хорошо согласуются и дополняют положения модели усредненного поля.
Известно, что при отсутствии модификатора полимер и глина термодинамически несовместимы и при их соединении происходит фазовое разделение. Избыточное количество модификатора, так же как и его отсутствие, затрудняет проникновение полимера в межслоевое пространство.
Согласно проведенным расчетам, увеличение длины молекулы полимера способствует фазовому разделению компонентов, поскольку энтропия длинных молекул при интеркаляции в узкий зазор уменьшается в большей степени, чем более коротких. Увеличение длины цепи модификатора, наоборот, способствует формированию термодинамически стабильного нанокомпозита. В этом случае наблюдается лучшее взаимопроникновение молекул полимера и модификатора, что увеличивает конфигурационную свободу обоих компонентов.
Результаты, полученные в обоих подходах, позволяют лучше понять факторы, определяющие способность полимера проникать в межплоскостные пространства слоистых силикатов и могут служить теоретической основой по практическому получению полимер-силикатных нанокомпозитов.
1.5.1. Выбор метода и оптимальных условий получения нанокомпозитов
При создании нанокомпозитов на основе слоистых силикатов метод получения выбирают исходя, прежде всего, из таких свойств полимера, как способность растворяться в растворителях, в которых набухает глина (растворное смешение), или находиться в вязкотекучем состоянии выше температуры стеклования или плавления (расплавное смешение).
Как уже отмечалось, способность полимера интеркалироваться в межплоскостные пространства зависит от парных взаимодействий полимер-глина, полимер-модификатор, глина-модификатор и изменения энтропии системы в целом.
Проникновение полимерных молекул в межплоскостное пространство глины определяется также и условиями проведения процесса. Часто для эксфолиирования слоистых силикатов в неполярных полимерах кроме модификаторов глины используют сомодификаторы, имеющие полярные группы и, обычно, достаточно длинные алифатические «хвосты», способные проникать в полимерную матрицу.
Если невозможно получить нанокомпозит из готового полимера проводят полимеризацию его мономера непосредственно в присутствии наполнителя. Полярные мономеры, способные спонтанно проникать в межплоскостное пространство глины, в результате полимеризации образуют интеркаляционные нанокомпозиты (ИН).
Наиболее сложная ситуация — получение ИН или в пределе ЭН на основе неполярных полимеров, например, полиолефинов без применения сомодификаторов. В этом случае приходится проводить процесс получения композита в термодинамически неравновесных условиях.
Во множестве публикаций показано, что в равновесных условиях, например, при получении композитов Na-ММТ/ПЭ методом адсорбции (удаление растворителя) [61-63] или при выдерживании смеси модифицированный Na-ММТ/полистирол (ПС) под давлением в расплаве при температуре выше точки размягчения [64], макромолекулы неспособны интеркалировать самопроизвольно в межплоскостные пространства глины независимо от типа используемого модификатора.
Увеличение полярности ПС хлорированием ароматического кольца привело лишь к незначительной интеркаляции цепей полимера в глину [64]. ИН на основе полипропилена (ПП) не образуются в результате смешения в расплаве с глиной, модифицированной аминами различной структуры. Для достижения совместимости предварительно модифицированной глины используют или функционализированный полярными группами ПП или в смесь глина/ПП добавляют некоторое количество сомодификатора. Обычно сомодификатором является функционализированный низкомолекулярный ПП [65-75]. В результате, в зависимости от соотношения глина/сомодификатор или, когда сомодификатор не применяют, от содержания полярных групп в ПП, получают ИН или ЭН.
1.6. Формирование нанокомпозитов в растворах полимеров на стадии полимеризации (поликонденсации)
Поиск и исследование саморегулирующихся систем, в которых одновременно протекают синтез полимерной матрицы и процесс образования нанокомпозита, может стать наилучшим решением задачи стабилизации наночастиц полимерами и их структурной организации.
Другими словами, речь идет о разработке методов создания нанокомпозитов с архитектурой «микрокапсулированная наночастица в полимерной оболочке», образованной in situ. Это осуществляется генерированием в полимеризующихся матрицах кластерных дисперсий, тем самым ограничивающих рост наночастиц. Пути для этого могут быть самые разные: полимеризация винильных мономеров в ходе интенсивного механического диспергирования оксидов металлов, введение в полимеризующуюся систему металлорганических соединений, которые разлагаются при температуре, близкой к температуре полимеризации, совместное γ-облучение прекурсора и мономера при комнатной температуре, полимеризация металлсодержащих мономеров и др. Толщина полимерной оболочки в таких нанокомпозитах регулируется концентрационными соотношениями и условиями полимеризации. Основные сложности на этом пути (особенно в случае длительных процессов отверждения эпоксидных, формальдегидных и других смол) — обеспечение седиментационной устойчивости в системах мономер—наночастица, а также необходимость учета внутренних напряжений, возникающих в таких системах.
Этот процесс представляет собой сложный многоступенчатый путь образования нанокомпозитов. Состав композита (долю отложенного вещества), как первый этап его исследования, определяют как с помощью химического анализа, так и по увеличению массы:
Δm = (mb − mp) / mp
где mb — масса полимера с наночастицами; mp — масса исходного полимера, которая может достигать в зависимости от исходных концентраций от долей до сотен процентов.
Полярные полимеры, интеркаляция цепей которых является термодинамически более выгодной, самопроизвольно проникают в межплоскостные пространства глины. Это водорастворимые полимеры: поливиниловый спирт (ПВС) [74, 75], полиэтиленоксид (ПЭО) [75-79], поливинилпирролидон (ПВП) [80], полиакриловая кислота (ПАК) [80].
Такие нанокомпозиты могут быть получены при добавлении водных растворов полимеров к дисперсиям разбухших слоистых натриевых силикатов или в органических растворителях. Таким методом ПЭО был успешно интеркалирован в Na-MMT и гекторит из ацетонитрила [81].
Интеркалированные нанокомпозиты получают также при полимеризации полярных неводорастворимых полимеров эмульсионной свободно-радикальной полимеризацией (глина с метилметакрилатом [82] и стиролом [83]), смешением в диметилацетамиде модифицированного ММТ с прекурсором полиимида — полиаминовой кислотой [84]. После удаления растворителя, полученную наполненную органоглиной пленку полиаминовой кислоты, нагревали до 300 °C для проведения реакции имидизации. В полученных полимеризацией нанокомпозитах обычно содержится некоторое количество неэкстрагируемого полимера. По данным РСА в равновесных условиях увеличение межслоиного пространства происходит практически всегда только на монослой полимера.
В определённых условиях, когда отношение количества полимера к содержанию глины велико и полимеризация происходит в расплаве предполимера, дифрактограммы композитов демонстрируют отсутствие рефлексов типичных для интеркалированной структуры, что привело авторов к заключению об образовании эксфолиированной структуры [4].
Полимеризацией ε-капролактама с раскрытием цикла были получены нанокомпозиты, содержащие Na-MMT, модифицированный 12-аминолауриловой кислотой (12-ММТ) [85, 86]. Данные РСА и посвечивающей электронной микроскопии (ПЭМ) показали, что в зависимости от количества введенного 12-ММТ, образуется или эксфолиированная (при менее чем 15 % масс. 12-ММТ) или интеркалированная структура (от 15 до 70 % масс. 12-ММТ). Дальнейшие работы показали, что интеркаляционная полимеризация ε -капролактама может быть осуществлена без обязательного модифицирования поверхности Na-MMT для увеличения его органофильности. Такая система чувствительна к кислоте, используемой для промотирования интеркаляции в ε-капролактам. Из всех исследованных кислот только ортофосфорная кислота позволяет получить, действительно эксфолиированный нанокомпозит (ЭН), другие кислоты, имеют тенденцию промотировать формирование частично эксфолиированных — частично интеркалированных структур. Причины этого не выявлены.
Messersmith и Giannelis [87] смешивали модифицированную алкилированную глину в расплаве с ε-капролактоном. РСА образцов не выявил заметного увеличения межслоиного расстояния (13.6 А). Авторы предполагают, что мономер внедряется в пустоты между цепями аминолауриловой кислоты, таким образом, что расширения галерей не происходит. Полимеризацию проводили при 170°С в течение 48 часов. Данные РСА полученных композитов свидетельствуют о произошедшей эксфолиации глины. Авторы считают, что интеркаляция мономера происходит лишь на стадии нагревания при 170°С.
Полярные олигомеры, например эпоксидные смолы, отверждённые малеиновым ангидридом образуют с глиной истинно эксфолиированные нанокомпозиты [88].
ЭН были получены с использованием бифункциональных модификаторов глины алкиламинов и винилового спирта. Функциональные группы модификаторов закреплялись на поверхности глины в межплоскостных пространствах. Виниловые группы сополимеризовывались с этиленом с использованием катализатора МАО [89-92]. ММТ, модифицированный цетилтриметиламммоний бромидом дополнительно обрабатывали додециламином и тетраэтоксисиланом или тетраметоксисиланом, которые после гидролиза образуют наночастицы двуокиси кремния. Это привело к дополнительному увеличению межслоевого расстояния. При полимеризации этилена в среде толуола образовывались ЭН [93]. ЭН были получены на основе глины, модифицированной L -аминокислотами или сложным модификатором (L -аминокислота-цетилтриметиламмоний бромид), полимеризацией этилена или сополимеризацией этилена и 1-октена с использованием цирконоценнового катализатора [94] и с чистым Na-MMT на катализаторе MgCl2 /TiCl4 [95].
1.7 Влияние глины на структуру граничных слоев полимера
Известен эффект влияние поверхности на структуру полимерных слоев, формирующихся вблизи неё при адсорбции из раствора или при охлаждении расплава [13], Работ, специально посвященных исследованиям структуры полимерных слоев на глине или особенностям кристаллизации полимеров в присутствие Na-MMT в настоящее время нет. В лучшем случае в некоторых статьях приводят разрозненные данные о структуре формирующихся полимеров.
Цепи водорастворимого полимера ПЭО спонтанно интеркалируется из водного раствора в Na-MMT. После экстракции полимера из нанокомпозита на глине остаётся монослой макромолекул. Изучение конформации адсорбированных цепей с использованием двухмерного двухфотонного ядерного магнитного резонанса (ЯМР) на G, обогащенном ПЭО, показало, что около 90-95 % -ОС-СО- связей ПЭО имеют гош- конформацию [96].
Стерические ограничения в расположении цепи ПЭО в межслойном пространстве приводят к снижению температуры и энтальпии плавления по сравнению с блочным полимером, а при дальнейшей интеркаляции — вплоть до полного подавления кристаллизации [97].
В нанокомпозитах, полученых в результате эмульсионной свободно-радикальной полимеризации метиметакрилата после экстракции в межслойном пространстве остаются один или два адсорбированных слоя полимера. По данным анализа экстрагированных композитов методом ИК-Фурье-спектроскопии, цепи ПММА на поверхности слоистого силиката располагаться плоско. На термограммах не фиксируется процесс стеклования полимеров. Авторы считают, что движущей силой, заставляющей органические полимерные цепи плоско лежать на поверхности слоистого силиката является ион-дипольное взаимодействие [83]; как и в образцах глина/ПММА, на ДСК-термограммах экстрагированных нанокомпозитов глина/ПС, полученных эмульсионной свободно-радикальной полимеризацией, также не наблюдается стеклования. Это позволило авторам предположить, что макромолекулы ПС находятся в межплоскостном пространстве глины в растянутой форме. Закрепление цепей ПС в межслойном пространстве не может быть объяснено ион-дипольным взаимодействием аналогично тому, как это было сделано для ПММА. Поэтому авторы в данном случае предполагают ион-наведенные дипольные взаимодействия между глиной и ПС [83].
В нанокомпозитах поли-8-капролактама (ПКЛ) наблюдали [87] понижение температуры плавления при увеличении массовой доли слоистого
силиката. Авторы объясняют это формированием кристаллитов малых размеров, возможно образующихся на высокоразвитой поверхности частиц глины. У некоторых адсорбированных полимеров температура стеклования вообще не фиксируется. Обычно адсорбированные макромолекулы не экстрагируются с глинами.
Применение механического воздействия на интекаляцию полимера в глину описан в работе [99]. Авторы работы получали нанокомпозиты модифицированная глина/поливиниловый спирт в растворе. Предварительная обработка глины в растворе ультразвуком перед добавлением полимера позволила увеличить долю эксфолиированного наполнителя.
1.8 Свойства полимер — силикатных нанокомпозитов
Как уже отмечалось, слоистые силикаты являются перспективными нанонаполнителями, которые улучшают механические свойства ряда полимеров, в которых они были диспергированы. Полученные материалы демонстрируют при достаточно низком количестве наполнителя (обычно менее 5 % масс.) улучшение свойств по отношению к свойствам самой полимерной матрицы. К таким свойствам относятся повышенный модуль упругости Юнга, прочность, повышенная термостойкость, и др. при сохранении (или незначительном ухудшении) эластичности. Рассмотрим более подробно наиболее типичные характеристики нанокомпозитов, для которых наблюдаются значительные улучшения по сравнению с их микрокомпозитными аналогами.
К другим перспективным характеристикам полимер-силикатных нанокомпозитов следует отнести пониженную газопроницаемость, улучшенные тепловые и огнеупорные свойства, высокую ионную проводимость и более низкий коэффициент теплового расширения. Повышенные барьерные свойства нанокомпозиционных материалов обусловлены тем, что силикатные слои непроницаемы для молекул жидкости и газа. Поэтому коэффициент диффузии у нанокомпозитов глина-полимер уменьшается в несколько раз по сравнению с коэффициентом диффузии исходных полимеров. Увеличение размера силикатных пластин приводит к снижению проницаемости [84, 107-112]. Коэффициент термического расширения также существенно уменьшается при добавлении даже небольшого количества глины (2-3 %) к полимерной матрице, так как жесткие слои силиката плохо деформируются и препятствуют тепловому расширению связанного с ними полимера. Отмечено, что нанокомпозиты, содержащие глину, имеют более высокую температуру разложения, чем чистый полимер, и, следовательно, являются более термоустойчивыми [8].
Природа и процессы, происходящие при горении нанокомпозитов на основе полимеров и глин, подробно описаны в работах Blumstein А., Giannelis Е.Р., Gilman J.W. и др. [101-109]. В этих работах показано, что при содержании глины в полимерах около 5 % масс, наблюдается заметное снижение скорости горения, снижается тепловыделение и увеличивается зольность. Материалы на основе ПЭ не теряют форму, расплавленные образцы не растекаются.
Возможность регулирования электропроводности ПЭО исследована в работе [97]. Нанокомпозит, полученный интеркаляцией из расплава полиэтиленоксида (40 масс. %) в Li-монтмориллонит (60 масс. %), демонстрирует повышенную стабильность ионной электропроводности при более низких температурах по сравнению с обычной смесью ПЭО/ Li-монтмориллонит.
Такое улучшение свойств объясняется тем, что ПЭО не способен кристаллизоваться в интеркалированном состоянии, вследствие чего исчезают кристаллиты, имеющие непроводящую природу. Более высокая ионная электропроводность при комнатной температуре по сравнению с обычными электролитами делает эти нанокомпозиты перспективными электролитными материалами.
Композиционные материалы на основе монтмориллонита и водорастворимых полимеров обладают высокими сорбционными свойствами. Некоторые композиты могут при определенных условиях неограниченно набухать в воде и образовывать соединения включения [110]. При этом, способностью к сорбции обладает не только поверхность композита, но межслоевое пространство. Такие материалы, как показывают исследования[110], обладают органофильными сорбционными свойствами и хорошими адсорбционными характеристиками по отношению к тяжелым металлам и их метилированным производным.
1.9 Гуанидинсодержащие полимеры: синтез и свойства
Гуанидинсодержащие соединения широко распространены в природе и находят применение в качестве физиологически активных веществ: лекарств, антисептиков, фунгицидов, пестицидов. К ним относятся, например, аминокислота аргинин, фолиевая кислота, многочисленные белки и нуклеиновые кислоты. Гуанидиновая группировка служит активным началом многих лекарственных веществ (сульгин, исмелин, фарингосепт, аспирин) и антибиотиков (стрептомицин и другие).
Высокую биоцидную активность гуанидиновым соединениям придает несущий положительный заряд катион гуанидиния, обеспечивающий электрическое взаимодействие с микробной клеткой.
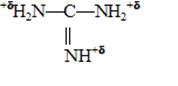
В отличие от четвертичных аммониевых соединений, где положительный заряд локализован на одном атоме азота, в катионе гуанидиния заряд распределен между тремя атомами азота. Такое строение реакционного центра обеспечивает необходимый баланс между эффективностью биоцидного действия антисептика в отношении микроорганизмов и токсичностью в отношении теплокровных животных и человека.
Макромолекулярная природа полигуанидинов обеспечивает пролонгированное антимикробное действие препарата: в отличие от низкомомкулярных соединений, антимикробное действие которых сохраняется всего несколько часов (в лучшем случае несколько суток), полимер образует на поверхности биоцидную пленку, которая обеспечивают длительную (несколько месяцев) защиту обработанной поверхности от появления на ней микроорганизмов. Наличие тонкой пленки, образующейся при протирании поверхности раствором 0,1 —1 % концентрации полигуанидина, было экспериментально подтверждено методом рентгеновской фотоэлектронной спектроскопии (РФЭС). Обнаружено, что полимерная пленка сохраняется на обработанной поверхности в течение нескольких месяцев и даже через 6 месяцев сохраняет биоцидную активность [111 ].
Комплекс свойств полигуанидинов позволяет использовать их не только в качестве антисептических средств в медицинской практике, но также в качестве биоцидных добавок в различные материалы (цемент, резину, ткани, бумажную массу, краски и др.), а также вспомогательных материалов в различных технологических процессах.
Первые данные о биоцидных свойствах гуанидиновых производных и полимеров на их основе были опубликованы в патентной литературе [112-113]. В указанных патентах описывается применение подобных соединений в качестве инсектицидов и отмечается, что соответствующие соединения особенно активны против грибковых заболеваний на фруктовых деревьях. В патенте [114] описаны специфические гуанидированные полиамины для применения против патогенных микроорганизмов.
К наиболее сильным из известных гуанидиновых антисептиков относятся «хлоргексидин» (1,6-бис-4,4-хлорфеноксибигуанидогексин) [110], низкомолекулярный полигексаметиленбигуанидин – «вантоцил» [115,116] и «космоцил» [117].
Так, например, хлоргексидин используется в качестве дезинфицирующего средства в виде солей (гидрохлорида, ацетата, глюконата) и широко рекомендуется за рубежом в виде растворов, мазей, присыпок как эффективное дезинфицирующее средство в хирургии для борьбы с внутрибольничными инфекциями, лечения кожных заболеваний и бытовых целей. Однако, следует отметить, что хлоргексидин, вантоцил и космоцил получают по сложной 4-х стадийной технологической схеме, кроме того при их синтезе исходным сырьем служит хлорциан, поэтому технологический процесс дорог и опасен.
В нашей стране был разработан процесс производства полимерного гуанидинового антисептика – полигексаметиленгуанидингидрохлорида (ПГМГ) («метацид», «полисепт») [118, 119], исходя из гексаметилендиамина и гидрохлорида гуанидина:
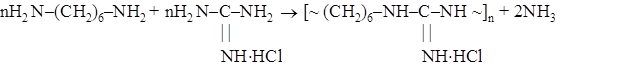
В дальнейшем было предложено объединение в одном процессе синтез гуанидингидрохлорида и получение из него ПГМГ [120].
Так как три аминогруппы гуанидингидрохлорида имеют различную реакционную способность, то молекулярную массу и структуру «полисепта» удается регулировать, изменяя условия реакции и содержание гексаметилендиамина в исходной смеси [121]. Так, при сравнительно низких температурах для процесса поликонденсации (120-130°С) в реакцию с гексаметилендиамином вступают преимущественно две аминогруппы гуанидингидрохлорида, образуя хорошо растворимый линейный олигомер с ММ 1,7-12,5´103. При увеличении количества гексаметилендиамина в реакционной смеси сверх одного моля на 1 моль гуанидингидрохлорида и повышении температуры до 180-200°С в реакцию может вступать третья аминогруппа и образуется разветвленный полимер, который имеет ММ 20 — 43´103 .
Различные соли ПГМГ (фосфат, глюконат, дегидроцет, сорбат, фторид, сульфат, нитрат, силикат, ацетат, стеарат, олеат, фумарат, сукцинат, адипинат, себацинат) были получены при действии различных кислот или их солей на основание или карбонат ПГМГ [122]. Среди указанных полимерных солей наибольшее практическое значение помимо «полисепта» имеют фосфат «фогуцид» и глюконат. По сравнению с «полисептом», фогуцид менее токсичен и коррозионноактивен.
Поликонденсационные полимеры: «полисепт» и «фогуцид» рекомендованы Минздравом в качестве дезинфицирующего средства в лечебных учреждениях и роддомах [123], а также в ветеринарии [124].
По данным указанным в работе [125, 126] растворы «полисепта» в концентрации 0,1-0,05% вызывают гибель грамположительных и грамотрицательных микроорганизмов: коринебактерий дифтерии (c. Duphtheretiae), золотистого стафилококка (St.aureus), а также St.aibus и St. faekalis, брюшно-тифозной палочки (S.typhi), шигелл Зонне и Флекснера (Shigella Sonnae, Flexneri), кишечной палочки (E.Coli), сальмонелл Бреслау и Гертнера (Salmonella th.murum), вульгарного протея (Proteus Vulgarus), синегнойной палочки (Ps.aeruginosa) в течение 5-25 минут.
В лаборатории поверхностно-активных полимеров и полиэлектролитов ИНХС РАН были синтезированы гуанидинсодержащие мономерные соли на основе дииаллилгуанидина (ДАГ) и органических кислот – уксусной и трифторуксусной (схема 1,2).
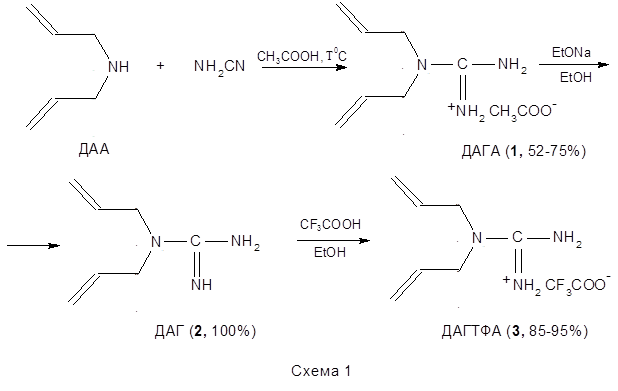

Проведенные исследования показали, что для данных мономеров скорость полимеризации невысока, и в результате реакции образуются полимерные продукты с невысокими молекулярными массами, что было подтверждено методами ЯМР1 Н. То есть, в рассматриваемом случае, несмотря на присутствие в качестве противоиона сильной ТФУК, не происходит, по-видимому, достаточного упрочнения связи α-протона диаллильной группы и вероятность его отрыва, а следовательно и ДПЦ на мономер высоки [127 ]. Такое поведение синтезированных мономеров ДАГА и ДАГТФА было объяснено следующим образом. В исследованных системах катионогенные мономерные соли ДАГА и ДАГТФА в водных растворах могут существовать в виде трех резонансных структур (I-III), из которых лишь одна отвечает необходимому для подавления деградационной передачи цепи на мономер условию — наличию заряда на азоте, связанного с двумя аллильными группами (структура I) (схема 3а).
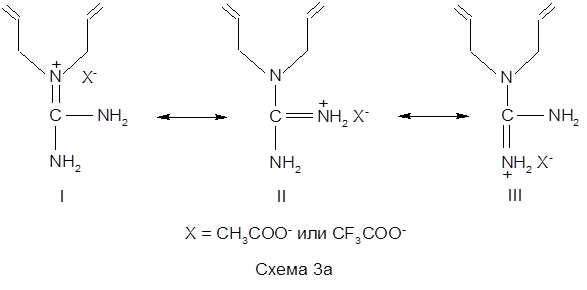
Тогда как структуры II и III, скорее всего, образуют стабильные (в том числе и по пространственным факторам) делокализованные системы с участием двух атомов азота и ацетатного (или трифторацетатного) противоиона (IV), либо за счет водородного связывания заряженной аминогруппы делокализованным карбоксилат-ионом (V) (схема 3б)
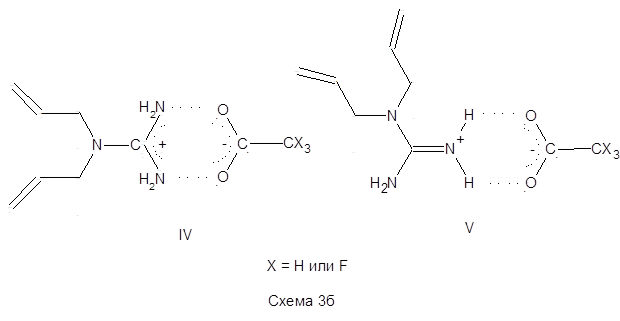
На основании этих предположений был сделан вывод, что при полимеризации мономерных солей (1) и (3) будет в значительной степени сохраняться деградационная передачи цепи на мономер. Для случая X = F (схема 3б) можно было бы ожидать снижения степени деградационной передачи цепи на мономер, так как высокий индукционный эффект (-I) трифторметильной группы должен был бы приводить к снижению устойчивости структур IV и V. Однако ожидаемого эффекта и заметных скоростей полимеризации ДАГТФА не удалось достичь, возможно, как предполагают авторы, еще из-за недостаточной растворимости мономера в исследованных системах [127].
В работах [127] исследовали радикальную полимеризацию мономерных солей метакриловой и акриловой кислоты и гуанидина (МАГ и АГ) и выявлены характерные особенности и кинетические закономерности этих процессов. Показано, что полимерные гуанидинсодержащие соли на основе ненасыщенных кислот способны полимеризоваться по радикальному механизму с образованием высокомолекулярных продуктов с биоцидными свойствами.
Никашиной с сотр.[128] разработан биоцидный сорбент клиноцид, представляющий собой природный цеолит (клиноптилолит-содержащий туф), на поверхности которого с помощью эпихлоргидрина закреплен полигексаметиленгидрохлорид. Клиноцид обладает катионо-обменной емкостью (1.0-1.5 мг-экв/мл), анионообменной емкостью (0.2-0.3 мг-экв/мл) и биоцидными свойствами; при прохождении через колонку с клиноцидом вода одновременно обессоливается и освобождается от бактериального и вирусного загрязнения (эффективность очистки 99-100%).
Таким образом, высокая биоцидная активность полигуанидинов в сочетании с низкой токсичностью, простотой синтеза и доступностью исходных веществ открывает возможность получения на их основе различных композиционных материалов, что расширит области их практического применения.
ГЛАВА 2. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
2.1 Подготовка исходных реагентов и растворителей
Гидроксид натрия, хлорид натрия, серная кислота марки «х.ч.»
Гидрохлорид гуанидина (ГГХ) «х.ч».
Метакрилат гуанидина (МАГ) «х.ч».
Акрилат гуанидина (МАГ) «х.ч».
Инертный газ- азот (о.с.ч).
Во всех опытах использована бидистиллированная вода, рН=7.
Очистка инициаторов.
Персульфат аммония (NH4 )2 S2 O8 (ПСА) “ч.д.а.” перекристаллизовывали из бидистиллированной воды несколько раз, затем высушивали в вакууме до постоянного веса.
Монтмориллонит месторождения Герпегеж с катионнообменной емкостью 95 мг-экв/100 г глины
Свойства мономеров-органомомодификаторов*
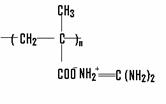
Вещество | Формула | Внешний вид | ММ | Растворимость | Тпл. |
Метакрилат гуанидина |
| Белый кр. пор. | 145,16 | Н2 O, CH3 OH, C2 H5 OH, C3 H8 OH, ацетон | 168 |
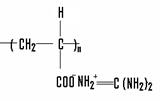
Акрилат гуанидина |
| Белый кр. пор. | 131.13 | Н2 O, CH3 OH, C2 H5 OH, C3 H8 OH, ацетон | 172 |
* Данные взяты из литературы [127].
2.2 Методика органомодификации бентонитовой глины месторождения «Герпегеж»
Получение органоглины из глинистого минерала осуществляли в две стадии. Первая стадия представляла собой операцию по концентрированию монтмориллонита путем удаления из глины балластных веществ, а вторая — перевод глинистого минерала в натриевую форму, удобную для получения органоглины.
Для концентрирования монтмориллонита применяли обычное отмучивание, т.е. промывание дистиллированной водой и удаление кальцита путем перевода его в хлорид кальция, обрабатывая золь глины 10%-ным раствором соляной кислоты. Причем нейтрализация кислотой золя глины осуществляли до тех пор, пока не прекратится образование пузырьков углекислого газа. Соляную кислоту в золь добавляли малыми порциями, так чтобы после прекращения реакции значение рН было не менее 3. Далее глинистый минерал промывали водой многократной декантацией.
Степень очистки от балластных веществ определяли по результатам ИК-спектроскопии по исчезновению пиков, относимых к кальциту.
Натриевая форма глины синтезирована при обработке золя (в которой отсутствуют балластные вещества) 1 М раствором хлорида натрия и через трое суток проводили отмывку глины от избытка хлорида натрия декантацией водой.
Полученный золь натриевой формы глины был исходным реагентом для преобразования в органоглину.
В качестве поверхностно-активных веществ – гидрофобизаторов нами были использованы гуанидинсодержащие мономеры- метакрилат и акрилат гуанидина.
Сначала приготавливали суспензию бентонита в воде путем перемешивания на магнитной мешалке в течение 2 часов. Затем к суспензии добавляли мономеры и перемешивали еще 6 часов.
2.3 Методика синтеза полимерных нанокомпозитов
К суспензии органомодифицированного гуанидинсодержащими мономерами бентонита в воде добавляли радикальный инициатор персульфат аммония в количестве 0,005 моль/л и перемешивали 30 минут. После этого суспензию продували азотом, разливали по ампулам со шлифами, изолировали от воздуха стеклянными пробками. Полимеризацию проводили в течение 8 ч при 60°С. Затем ампулы разбивали, извлекали полученные композиты, промывали дистиллированной водой и помещали в избыток дистиллированной воды на сутки.
2.4 Методы исследования и методики эксперимента
2.4.1 ИК-спетроскопическое исследование исходных соединений и синтезированных продуктов
Сканирование ИК-спектров осуществлялось на ИК Фурье-спектрометре ФСМ 1201 с компьютерной системой управления, производства АО «СПб Инструментс» (Санкт-Петербург). Частотная область исследования 400-5000 см-1. Режим работы прибора: разрешение – 4 см-1, число сканов –10, отношение сигнал/шум >1300.
2.4.1.1 Методика подготовки образцов для сканирования ИК-спектров
Твердые образцы для сканирования их спектров поглощения подготавливаются двумя способами: в виде суспензии в вазелиновом масле, либо в виде таблеток в бромиде калия.
Приготовление суспензии веществ в вазелиновом масле
Суспензию приготавливают путем измельчения и растирания анализируемого вещества в вазелиновом или другом минеральном масле. Для растирания используют ступку из яшмы (агата).
В ступку помещали 5-10 мг исследуемого вещества, затем при помощи пипетки наносили каплю масла на середину головки пестика и начинали энергично растирать им вещество. Сделав пестиком около пятнадцати круговых движений, с помощью шпателя из нержавеющей стали (при его отсутствии можно использовать полоски бумаги) собирали всю суспензию в центре ступки и повторяли растирание.
Приготовление суспензии считали законченным после трехкратного повторения операций. Приготовленную суспензию наносили на центральную часть окошка из КВr и придавливали сверху вторым окошком, добиваясь равномерного растекания пленки из суспензии по поверхности.
Толщину пленки выбирали с таким расчетом, чтобы наиболее интенсивной полосе в исследуемой области спектра соответствовало пропускание около 5 %.
При изготовлении образцов методом прессования таблеток в качестве вещества матрицы использовали чистый КВr, не содержащий посторонних полос поглощения, предварительно размельченный и осушенный.
Прессование таблеток осуществляли в следующей последовательности. С помощью аналитических весов отвешивали 1 ÷ 2 мг исследуемого вещества, к которому затем добавляли порошок КВr, доводя общий вес навески до 300 мг. В зависимости от интенсивности полученного спектра количества исследуемого вещества увеличивали или уменьшали.
Приготовленную навеску загружали в полушарие вибромельницы и измельчали в течение 1-2 мин. Измельченную смесь исследуемого вещества и КВr равномерным слоем засыпали в пресс-форму поверх нижнего пуансона и слегка утрамбовывали с помощью верхнего пуансона, вращая его рукой.
Собранную пресс-форму подсоединяли к форвакуумному насосу и помещали в гидравлический пресс. Части пресс-формы поджимали для создания должного уплотнения резиновыми прокладками.
Пресс-форму откачивали в течение 5-10 минут для удаления из пробы воздуха. Не прекращая откачки, прикладывают к пресс-форме необходимое для прессования таблетки усилие 8,0 т/см2, и производят прессование таблетки в течение 2 мин. При общем весе засыпаемого вещества 300 мг в пресс-форме диаметром 13 мм получалась таблетка толщиной около 1 мм.
Полученные таким образом таблетки не растрескивались и оставались прозрачными в течении нескольких часов.
2.4.2 Термофизические методы исследования
Термофизические методы исследования. Фазовое поведение систем изучали на термоаналитической установке (МОМ, Венгрия) с нагревательной ячейкой DSС 30 при скорости нагревания 5 град× мин-1 в саморегенерирующейся атмосфере воздуха.
Термическую устойчивость изучали методом дифференциального термического анализа (ДТА) и дифференциальной термической гравиметрии (ДТГ) на дериватографе 1000D (фирма МОМ, Венгрия) на воздухе при скорости нагревания 2.5 град× мин -1 от 20 до 5000С.
Термофизические данные обрабатывались с использованием программно-аппаратного комплекса для измерения сигналов дериватографа 1000D (фирма МОМ, Венгрия) и компьютерной обработки данных термогравиметрического анализа в Институте химии растворов РАН (г. Иваново).
2.4.3 Рентгеноструктурный анализ
Дифракционные данные получены при комнатной температуре на автоматизированном дифрактометре ДРОН-6 (36кВ, 20мА, λСиКα, графитовый монохроматор на вторичном пучке, съемка по Бреггу-Брентано в интервале углов 2θ от 1 до 30°, шаг 0,05°, скорость сканирования 1град/мин).
4.4. Исследование удельной поверхности и распределения частиц по размерам.
Удельную поверхность композитов и распределение частиц по размерам исследовали на лазерном анализаторе частиц “MicroSizer 201”. Ультразвук=200 W, время диспергирования 60 сек., коэффициент пропускания=79.
2.5 Исследование бактерицидной активности композиционных материалов
Для изучения бактерицидной активности синтезированных мономеров и полимеров в качестве тест-микробов использованы штаммы кишечной палочки E-coli и стафилоккока St.aureus из международной коллекции эталонных штаммов, микробная нагрузка которых составляла 0,1 мл 1 миллиардной суспензии на 1 мл препарата, использованного в определенных концентрациях (двукратные разведения препарата в стерильном растворе дистиллированной воды). В контрольную пробирку препараты не добавляли, в них содержались тест-штаммы E-coli, St.aureus и 1 мл.дистиллированной воды. Экспозиция препаратов с тест-микробами составляла 1 час при комнатной температуре. Далее проводился высев из каждой опытной пробирки и из контрольной на питательный агар Эндо, разлитый в стерильные чашки Петри. Затем чашки с посевами инкубировали в термостате при 37 °С в течение 18 часов и определяли антибактериальную активность изучаемых образцов.
Эффективность бактерицидного действия композитов определяли по количеству колониеобразующих единиц (КОЕ), рассчитывая их жизнеспособность по формуле: С=lg Nt /Ntk, где Nt – количество бактерий, вышивших после обработки композитом, Ntk — количество бактерий в контроле за один и тот же промежуток времени.
2.6 Методика определения адсорбционных свойств композиционных материалов
О сорбционных свойствах глин обычно судят по обесцвечиванию водных растворов красителей. В качестве красителя используется водорастворимый краситель метиленовый синий (голубой). Этот краситель имеет химическое название 3,7-бис (диметиламино) – фетазин.
Метиленовый синий широко применяется в биологическом и химическом анализах. Краситель растворим в воде, плохо растворим в этаноле. В практике он используется в виде 0,10%-ного водного раствора. Для его приготовления 0,10 г красителя переносят количественно в мерную колбу на 100 мл и разбавляют водой до метки.
Для того, чтобы сравнить сорбционные возможности глины необходимо провести количественные измерения и построить кривую адсорбции.
Для этого в пробирки вливают по 50 мл рабочего раствора красителя, затем засыпают 0,4; 0,5; 0,6; 0,7; 0,8; 0,9 г глины. Пробирки с раствором взбалтывают и оставляют на сутки, затем фильтруют отдельно содержимое каждой колбы через бумажные фильтры. Отбирают 1 мл отфильтрованного раствора, помещают в кювету, устанавливают длину волны λ=750 нм и фотометрируют.
Равновесную массу метиленового синего в растворе находят по градуированному графику. Для построения градуировочного графика отбирают 40 мл приготовленного рабочего раствора в мерную колбу объемом 200 мл с С=0,2 мг/мл и доводят до метки дистиллированной водой. Затем в мерные колбы на 50 мл вливают 1,2,3,4,5,6 мл этого раствора красителя и доводят до метки водой.
На основании табличных данных определяют lg х/m. И строят изотерму адсорбции исходя из подчинения уравнению Фрейндлиха в логарифмической форме: ![]()
Рис. Градуировочный график определения метиленового синего
2.6.1 Количественное определение фенола по методу Коппешара
Техника безопасности при работе с фенолами
Фенол – токсичное вещество. При попадании на кожу он разъедает ее и вызывает ожог, с последующим образованием язв. При попадании фенола на кожу необходимо сразу же смыть ее сильной струей воды, а затем обработать 96 % этанолом, содержащим 3% хлорного железа, и, наконец, снова водой. В более серьезных случаях поражения фенолом или его парами необходимо немедленно обратиться к врачу.
Взвешивание фенола необходимо производить в вытяжном шкафу. Предельно-допустимая концентрация фенола в воздухе лаборатории составляет 5 мг/см3 .
2.6.1.1 Количественное определение фенола
Свободный фенол определяют, обрабатывая его бромид-броматным раствором в кислой среде. Вначале образуется трибромфенол (трибромкрезол). При избытке брома бромирование идет глубже с образованием конечного продукта реакции трибромфенолброма (трибромкрезолброма). При добавлении к реакционной смеси раствора иодистого калия избыток брома и бром, замещающий водород гидроксильной группы, вытесняют эквивалентное количество иода. Выделяющийся йод титруют 0,1 н. раствором тиосульфата натрия.
Методика эксперимента
Приготовление модельного раствора фенола
Навеску фенола 2 – 3 г, взятую на аналитических весах по разности, помещают в мерную колбу емкостью 500 мл, растворяют в 20 мл метилового или этилового спирта и добавляют воды до метки. Для определения фенола 25 мл раствора вносят пипеткой в коническую колбу емкостью 250 мл с притертой пробкой, прибавляют 50 мл бромид-броматного раствора, выдерживают колбу со смесью во льду 10 мин, затем прибавляют 5 мл соляной кислоты (плотность 1,19 г/см3 ), закрывают колбу пробкой, взбалтывают смесь и через 15 минут титруют выделившийся иод 0,1н. раствором тиосульфата натрия. В конце титрования (когда раствор становится светло-желтым) прибавляют 1 мл раствора крахмала в качестве индикатора и продолжают титровать до обесцвечивания раствора. Параллельно проводят контрольный опыт.
Содержание фенола рассчитывали по формуле:
X = 500 (υ1 − υ2 ) FK 100 = 2000 (υ1 − υ2 ) FK / g,
25g
где υ1 – объем 0,1 н раствора тиосульфата натрия, пошедшего на титрование в контрольном опыте, мл; υ2 — объем 0,1 н раствора тиосульфата натрия, пошедшего на титрование испытуемого раствора, мл; F –количество фенола, соответствующее 1мл 0,1 н. раствора тиосульфата натрия, г (для фенола F равно 0,001567); K-поправка к титру; g- навеска фенола, г.
2.6.2 Определение тяжелых металлов атомно-абсорбционным методом
Тяжелые металлы в пробах воды до и после очистки определяли атомно-абсорбционным методом с электротермической атомизацией с использованием атомно-абсорбционного спектрометра „МГА-915“. Содержание металлов определялось величиной интегрального аналитического сигнала и рассчитывалось по предварительно установленной градуировочной зависимости.
2.7 Методика определения степени набухаемости и плотности композитов
Для определения степени набухаемости навеску полимерного композита помещали в дистиллированную воду и растворы мочевины и фурациллина и оставляли на ночь. Затем набухшие композиты извлекали из раствора и взвешивали.
Степень набухания полимерных композитов F характеризовали отношением массы композита, набухшего в воде, mw к массе сухого композита md :
F = mw / md
Плотность композитов измеряли пикнометрическим методом.
ГЛАВА 3. ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
3.1 Синтез новых композиционных материалов на основе монтмориллонита и гуанидинсодержащих полимеров
Нанокомпозиты на основе водорастворимых полимеров и слоистых силикатов представляют собой новый и очень перспективный класс материалов. Основной особенностью минерально-органических материалов является то, что в них сочетаются свойства исходного неорганического материала и органического полимера. Используя различные сочетания минерала и полимера, можно варьировать свойства данной системы в широких пределах в зависимости от их дальнейшего использования. Одной из наиболее перспективных областей применения минерально-органических композиционных материалов является ионообменная и сорбционная очистка воды. Для этих целей определенный интерес представляют слоистые силикаты, модифицированные гуанидинсодержащими полимерами. Как известно, соединения, содержащие гуанидиновые группы, проявляют заметную биоцидность [111]. Введение таких групп в полимерные цепи приводит к усилению этих свойств, поэтому создание новых полимеров, содержащих гуанидиновые группы, изучение их физико-химических свойств и характеристик, а также выявление возможности получения на их основе нанокомпозиционных полимерных продуктов, обладающих биоцидными свойствами, представляется нам весьма важным направлением исследований.
Анализ литературных данных (см. лит. обзор) показал, что исследования структуры и физико-механических свойств перспективных нанокомпозитов на основе водорастворимых ионогенных полимеров и глин находятся на начальной стадии и мало изучены.
Разработка методов получения, в результате которых образуются нанокомпозиты с различной степенью интеркалирования полимера в слоистый силикат вплоть до полной эксфолиации наполнителя, является в настоящее время важнейшим этапом разработки фундаментальных основ создания нанокомпозитов. Наконец, использование в качестве полимерной матрицы водорастворимых ионогенных полимеров делает получение интеркалированных или эксфолиированных полимер-силикатных нанокомпозитов самостоятельной задачей в данном исследовании.
Сам метод получения композита влияет на структуру всех компонентов материала. Один из перспективных путей получения полимерных нанокомпозитов – радикальная полимеризация и сополимеризация мономеров ионизующихся в водных растворах в присутствии слоистых силикатов, например монтмориллонита.
В данной диссертации исследован способ полимеризационной модификации монтмориллонита в водных растворах с применением полифункциональных гуанидинсодержащих мономерных систем, а также изучено влияние комплекса физико-химических свойств полимерных нанокомпозитных материалов на их сорбционные и биоцидные характеристики.
Для решения поставленных задач в качестве исходных объектов для получения новых полимерных нанокомпозитов были выбраны гуанидинсодержащие мономеры винилового ряда метакрилат (МАГ) и акрилат (АГ) гуанидина. Выбор этих мономеров обусловлен совокупностью причин: во-первых, полимеры на основе мономеров акрилового ряда (ПАК и ПМАК) являясь полиэлектролитами, обладают, несомненно, широким спектром практически полезных свойств и применяются, как известно, в самых разнообразных отраслях промышленности, а во-вторых, обладают собственной биоцидной активностью, в случае мономеров акрилового ряда последнее относится к соответствующим солям.
Как уже сообщалось в обзоре литературы, получение нанокомпозитов на основе полимеров и природных алюмосиликатов связано с принципиальными трудностями вследствие несовместимости органической фазы полимера и неорганической природного минерала. Для улучшения совместимости полимерной матрицы с монтмориллонитом необходима дополнительная «модификация» глинистых минералов, а именно, гидрофобизация поверхности глины обработкой поверхностно-активными алкиламмониевыми солями. Именно наличие положительного заряда на атоме азота алкиламмониевых солей является ключевым фактором для решения рассматриваемой проблемы. Четвертичные алкиламмониевые катионы могут вытеснять ионы Na+ с обменных позиций в монтмориллоните и понизить поверхностную энергию глины так, что между слоями глины могут внедряться органические молекулы с различной полярностью.
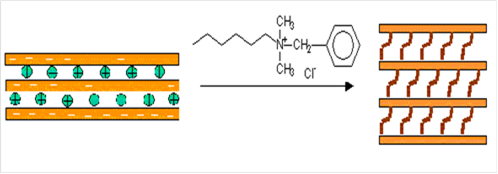
Рис. 3.1. Схема органической модификации монтмориллонита
Модифицированная в результате хемосорбции глина (или органоглина) являясь органофильной, имеет меньшую поверхностную энергию и лучше совмещается с органическими полимерами. Когда межплоскостные катионы после ионного обмена замещены более объемными алкиламмониевыми органическими катионами, происходит также и увеличение межслоевого расстояния. Эти свойства позволяют применять органоглины для получения нанокомпозитов с участием полимеров различной природы.
В представленной работе в качестве поверхностно-активных веществ для модификации поверхности глины были использованы гуанидинсодержащие мономеры винильного ряда метакрилат и акрилат гуанидина, синтезированные в работе [112].Нами было сделано предположение, что наличие в структуре гуанидинсодержащих мономеров положительно заряженного атома азота открывает возможность их применения в качестве гидрофобизаторов поверхности монтмориллонита.
В качестве глинистого минерала был использован монтмориллонит месторождения «Герпегеж» Кабардино-Балкарской республики.
Бентонитовая глина месторождения «Герпегеж» является многокомпонентной системой, в которой доля кальциевого монтмориллонита по нашим данным, полученным методом ИК-спектроскопии, составляет 15-20%, а массовая доля балластных веществ, первую очередь кальцита более 50%. Поэтому получение органоглины из глинистого минерала осуществляли в две стадии. Первая стадия представляла собой операцию по концентрированию монтмориллонита путем удаления из глины балластных веществ, а вторая — перевод глинистого минерала в натриевую форму, удобную для получения органоглины.
Для концентрирования монтмориллонита можно было бы применить обычное отмучивание, однако отделить кальцит полностью нам не удалось. Не удается удалить кальцит из золя глины и центрифугированием.
Наиболее приемлемым способом концентрирования монтмориллонита в бентонитовой глине является удаление кальцита путем перевода его в хлорид кальция, обрабатывая золь глины 10%-ным раствором соляной кислоты:
СаСО3 + 2НС1 ® СаС12 + 2СО2 + Н2 О (1)
Причем нейтрализация кислотой золя глины осуществлялась до тех пор, пока не прекратилось образование пузырьков углекислого газа. Соляную кислоту в золь добавляли малыми порциями. После прекращения реакции значение рН было не менее 3. После такой обработки глины и промывки глины водой (декантацией), пики на ИК-спектре, относимые к кальциту исчезают.
Однако химическая обработка соляной кислотой может приводить к изменению природы глинистого минерала или появлению новых твердых фаз. На это указывают некоторые литературные источники, а в наших экспериментах результаты РФА. Так, было установлено, что полученная натриевая форма глины содержит некоторое количество фазы, представляющая гидросиликат кальция Ca5 (SiO4 )2 (OH)2 и которой на рентгенограмме соответствует интенсивный пик с межплоскостным расстоянием 3,346 Å.
Натриевая форма глины синтезирована путем обрабатывания золя глины (в которой отсутствуют балластные вещества) 1 М раствором хлорида натрия (3 суток) и последующей отмывкой глины от избытка хлорида натрия декантацией водой.
Нанокомпозиты на основе монтмориллонита и гуанидинсодержащих полимеров получали в процессе радикальной полимеризации мономеров в водном растворе суспензии глины в присутствии радикального инициатора персульфата аммония (см. экспериментальную часть).
В результате исследований было выявлено, что гуанидинсодержащие соединения являются хорошими модификаторами поверхности и межслоевого пространства монтмориллонита.
Для изучения степени распределения органомодифицированного монтмориллонита в нанокомпозите нами использовался метод рентгеноструктурного анализа. Результаты анализа показали: в случае монтмориллонита наблюдается характерный пик в области 2Q = 8,53° (d = 1,178 нм), отвечающий за расстояние между слоями глины; при введении гуанидинсодержащего полимера в межслоевое пространство пик смешается в малоугловую область, что свидетельствует о раздвижении слоев глины.
О внедрении мономеров в глинистые минералы судили также по результатам ИК-спектроскопии по наличию в спектрах композитов характеристических полос мономеров.
Были определены оптимальные условия проведения реакции радикальной полимеризации (температура полимеризации 60о С, интервал концентраций мономера 50-85 масс.%, концентрации инициатора (ПСА) 0,02 моль/л, концентрация монтмориллонита 15-50 масс.%, время 60 минут, в которых изучаемая реакция протекает с хорошим выходом.
Получены образцы нанокомпозитов с органомодифицированным монтмориллонитом как в нативной так и в натриевой форме. Полученные образцы представляют собой гелеобразные однородные массы, набухающие в воде.
Возникает вопрос: какова природа сил, вызывающих активную адсорбцию гуанидинсодержащих мономеров монтмориллонитом? Исходя из наличия в молекулах мономеров (схема 1) групп СО и NH можно ожидать их высокую способность к образованию водородных связей.
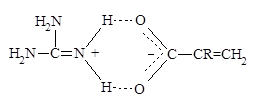
R=H, CH3 ,
Схема 1. Структура метакрилата и акрилата гуанидина
По-видимому, молекулы гуанидинсодержащих мономеров разрушают водородные связи между кристаллитами бентонита и внедряются между ними за счет образования новых связей. Это предположение подтверждается экспериментами, проведенными в работе [110] при исследовании степени набухания нанокомпозитов на основе акриламида и бентонитовой глины в растворах мочевины и фурацилина. Авторы обнаружили, что в растворах фурациллина композиты набухают значительно сильнее, что было объяснено наличием карбонильных и аминогрупп и большими размерами молекул фурациллина.
Для приготовления композитов использовали достаточно концентрированную суспензию бентонита. В этой области концентраций большую роль играют силы притяжения между ребрами и плоскостями кристаллитов. Суспензия приобретает структуру карточного домика, схематически изображенную на рис. 3.2. (а)
а) б)
б)

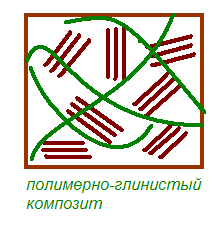
в) г)
Рис. 3.2. Структура суспензии глины и композитов
При механическом перемешивании происходит разрушение контакта типа край – плоскость, т.е. разрушение карточного домика (рис.3.2 в) [109-110]. Именно частично разрушенная структура фиксируется при полимеризации и образовании полимерной сетки (рис.3.2 г).

Рис.3.3. Схема образования слоистого нанокомпозита на основе алюмосиликата и полимера с низким его содержанием (справа вверху) и высоким.
Использование в качестве гидрофобизатора и модификатора в межслоевом пространстве глины гуанидинсодержащего мономера способного к дальнейшей полимеризации значительно упрощает методику получения полимерной композиции и уменьшает длительность процесса.
Изучение распределения органоглины в полимерной матрице имеет большое значение, так как свойства получаемых композитов напрямую зависят от степени распределения органоглины.
Согласно работам Джианелиса [63] процесс формирования нанокомпозита протекает через ряд промежуточных стадий. На первой стадии происходит образование тактоида – полимер окружает агломераты органоглины. На второй стадии происходит проникновение полимера в межслойное пространство органоглины, в результате чего происходит раздвижение слоев до 2-3 нм [98]. Дальнейшее увеличение расстояния между слоями (третья стадия) приводит к частичному расслоению и дезориентации слоев органоглины. Эксфолиация или расслоение наблюдается, когда полимер раздвигает слои глины на 8 – 10 нм и более.
На самом деле, в получаемых полимерных нанокомпозитах могут присутствовать все указанные структуры, что зависит от степени распределения органоглины в полимерной матрице. Расшелушенная (эксфолиированная) структура является результатом очень хорошей степени распределения органоглины.
Данные рентгеновских исследований гуанидинсодержащих композитов показали, что для композитов полное определение кристаллографических параметров затруднено, ввиду того, что в области углов 2Q=19.8-23.0° небазальные рефлексы глины экранируют пики полимера.
Поскольку в результате интеркаляции мономера в межпакетные промежутки силиката базальный рефлекс может сдвигаться в область истинно малых углов, для подробного анализа этого диапазона была получена дифрактограмма в малых углах дифракции для органоглины (глина, обработанная мономером) и одного из композитов с исходным составом 50:50 масс. %. В интервале углов 2Q от 1.0° до 2.0° рефлекс, соответствующий межплоскостному расстоянию монтмориллонита 1.5 нм, не был обнаружен для органоглины, а в композите зарегистрировано его наличие.
На основании проведенных исследований можно сделать вывод о том, что органоглина, полученная обработкой монтмориллонита гуанидинсодержащими мономерами, является полностью эксфолиированной структурой, а композиты, при небольших (примерно до 15 % масс.) степенях наполнения являются эксфолиированными, а при более высокой концентрации полимера — нанокомпозитами смешанного типа, которые содержат интеркалированные кристаллиты и эксфолиированные элементарные пакеты.
Таким образом, гуанидинсодержащие полимерные композиционные материалы были получены интеркалированием гуанидинсодержащих мономеров в слои глины и последующей их полимеризацией в присутствии радикальных инициаторов.
3.2 Особенности взаимодействия гуанидинсодержащих полимеров с глинистым минералом
Основными факторами, оказывающими влияние на процесс получения композита на основе монтмориллонита и виниловых мономеров являются концентрация мономера, инициатора, температура.
С увеличением содержания мономеров от 10 до 50 % количество привитого полимера увеличивается незначительно. Дальнейшее увеличение концентрации мономера также незначительно влияет на степень прививки даже при содержании 75 % мономера степень прививки не превышает 25 %. Видимо, при высоких концентрациях мономера в основном протекает реакция гомополимеризации. Авторы работы [4] отмечают также, что при проведении сополимеризации различных мономеров с силикагелями не получили значительного увеличения количества привитого полимера.
С увеличением концентрации инициатора от 0,02 до 0,06 моль/л, т.е. в пределах концентраций, обычно применяющихся в практике проведения радикальной полимеризации виниловых мономеров, количество привитого полимера возрастает. Однако, большое содержание инициатора > 0,04 моль/л приводит к снижению выхода конечного продукта, что вероятно, связано с трудностями, возникающими при адсорбции мономера на поверхности, содержащей избыток инициатора.
Результаты исследования продолжительности прививки мономера к бентониту показывают (табл.3.2.1.), что эффективность процесса модифицирования достигает максимальных значений за 60 минут.
Как видно из таблицы, поведение МАГ в реакциях полимеризации в присутствии монтмориллонита имеет черты отличные от поведения АГ в тех же условиях. Степень прививки МАГ к монтмориллониту выше.
Возможное объяснение большей реакционной способности МАГ заключается в стабильности образующего метакрилатного радикала, в котором осуществляется сверхсопряжение как с атомами водорода метильной группы, так и с карбонильным кислородом и гуанидиновой группой. Большая делокализация заряда карбоксильной группы в молекуле мономера МАГ подтверждается данными ЯМР1 Н-спектроскопии показывающими смещение сигналов винильных протонов МАГ в более сильное поле по сравнению с АГ [129].
По всей вероятности, наблюдаемые особенности и различия в ряду рассматриваемых мономеров объясняются сложным характером вкладов различных физико-химических процессов, определяющих протекание реакции радикальной полимеризации гуанидинсодержащих мономеров акрилового ряда на поверхности и в межслоевом пространстве глинистого минерала. Вместе с тем, основной вклад в изменение эффективной реакционной способности полимеризующихся частиц вносят, как мы полагаем, ассоциативные взаимодействия между гуанидиновыми и карбоксильными группами (как внутри- так и межмолекулярные) и структурная организация соответствующих мономеров и полимеров в процессе образования нанокомпозита.
Таблица 3.2.1
Результаты взаимодействия гуанидинсодержащих соединений с монтмориллонитом*
образец | Концентрация мономера в исходном растворе, % | Концентрация инициатора, моль/л | Время, мин | Количество привитого полимера, % |
АГ | 10 | 0,02 | 60 | 5,59 |
30 | 6,21 | |||
50 | 16,67 | |||
30 | 0,01 | 60 | 5,23 | |
0,02 | 6,21 | |||
0,04 | 13,42 | |||
0,06 | 6, 57 | |||
30 | 0,04 | 10 | 8,0 | |
20 | 8,4 | |||
30 | 9,6 | |||
60 | 13,42 | |||
МАГ | 10 | 0,02 | 60 | 6,2 |
30 | 7,4 | |||
50 | 18,6 | |||
30 | 0,01 | 60 | 6,1 | |
0,02 | 7,4 | |||
0,04 | 14,8 | |||
0,06 | 6,93 | |||
30 | 0,04 | 10 | 8,6 | |
20 | 9,4 | |||
30 | 10,2 | |||
60 | 14,8 |
* Примечание. Количество полимера связанного с монтмориллонитом определяли элементным анализом на азот.
3.3 ИК-спектральные исследования исходных и синтезированных продуктов
3.3.1 ИК– спектральный анализ бентонитовой глины месторождения Герпегеж
Глину месторождения Герпегеж подвергали всестороннему изучению методом ИК-спектроскопии. При этом анализировались изменения полос поглощения на спектрах этой глины после обработки кислотой (рисунок 13), обогащения некоторых фаз глины методом центрифугирования (рисунок 14) и переведения ее в натриевую форму. На рисунке 10а представлен ИК-спектр воздушно-сухого образца глины.
Содержание минерала кальцита в образцах глины Герпегеж подтверждается наличием в спектре полос в областях: 712 см-1, 874 см-1, 1431 см-1, 1796 см-1, 2513 см-1. Другая часть полос поглощения связана с монтмориллонитовой составляющей глины. Наблюдаемую интенсивную полосу поглощения с тремя ярко очерченными зубцами при 429, 468 и 519 см-1 можно отнести к деформационным колебаниям кислородо-кремниевых группировок типа Si-O-Si (429, 468 см-1 ) и Si-O-Al (519 см-1 ). Соответствующие валентные колебания этих группировок отражаются на инфракрасном спектре в виде сильной по интенсивности полосы 1037 см-1. Присутствие абсорбционной воды и гидроксильных группировок в составе глины подтверждается наличием деформационных колебаний ОН-групп молекул воды при 1625 см-1. Деформационные колебания гидроксильных групп проявляются в области 916 см-1. Валентное колебание ОН-групп молекул абсорбционной воды наблюдается в виде сильной полосы при 3439 см-1. Валентное колебание ОН-группы соответствует пику с волновым числом 3628 см-1 .
Вызывало сомнение отнесение полосы поглощения, представляющего дуплет при 779 и 798 см-1. С одной стороны этот дуплет является характеристическим признаком минерала монтмориллонитовой группы бейделита, с другой стороны этот дуплет может быть отнесен к поглощению кварца. Охарактеризовав полосы поглощения на ИК-спектре образца глины месторождения Герпегеж, можно, по-видимому, провести аналогичное отнесение полос поглощения на спектре глины. Действительно, полосы поглощения 713 см-1, 874 см-1, 1423 см-1, 1797 см-1, 2513 см-1 – позволяют идентифицировать в глине кальцит (СаСО3 ), что подтверждается рентгенофазовым анализом.
Особенностью ИК-спектра образца глины является слабая по интенсивности узкая полоса при 3699,8 см-1. По-видимому, это указывает на наличие в составе глины фазы, содержащей свободную ОН-группу.
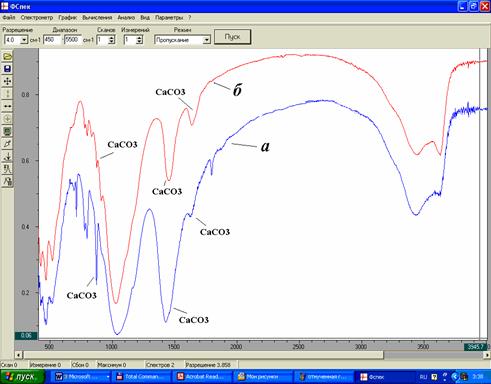
Рисунок 3.3.1. ИК-спектры бентонитовой глины Герпегеж: а — исходный образец глины; б — образец после 20 мин. центрифугирования (3000 об/мин)
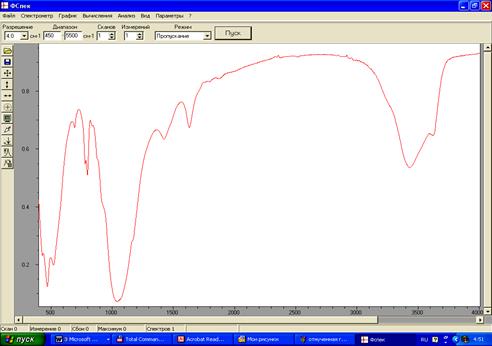
Рисунок 3.3.2. ИК-спектр образца бентонитовой глины после обработки 10%-ным раствором соляной кислоты
3.3.2 ИК-спектральные характеристики композиционного материала на основе монтмориллонита и гуанидинсодержащих полимеров
В данном разделе рассматриваются ИК-спектральные характеристики органоглины и композиционных материалов на основе монтмориллонита и гуанидинсодержащих полимеров. ИК спектры всех соединений регистрировали в твердом виде в таблетках KBr.
В качестве модельных соединений были взяты мономерные и полимерные соединения и бентонитовая глина, которые должны были подтвердить соответствующие изменения в спектрах при переходе от исходных веществ к композиционным материалам.
Образование нанокомпозита на основе монтмориллонита и гуанидинсодержащих полимеров сопровождается смещением всех характеристических полос поглощения минерала в высокочастотную область: так полоса поглощения 950 см-1 проявляется в области 1010 см-1, 1150 см-1 смещается в более слабое поле на 30 см-1 и проявляется при 1180 см-1, 520 см-1 при 560 см-1 .
Валентные колебания акрилатного компонента проявляются в области 1720 см-1. Полоса поглощения, характерная для деформационных колебаний молекул воды (1632 см-1 ), отчетливо проявляется в спектре модифицированного бентонита. Область при 1480 см-1 соответствует симметричным валентным колебаниям метиленовых групп.
Также в композитах в отличие от чистого бентонита появляются две широкие 3350 и 3100 см-1, которые относятся к валентным колебаниям N-H связей в гуанидиновом катионе.
Метакрилатный противоион метакрилата гуанидина проявляется в спектре в виде типичных полос, характеризующих карбоксилат-ион метакриловой кислоты. Так, широкая и очень интенсивная полоса 1530 см-1 относится к валентным колебаниям связи С=О в анионе – СОО-. Другие полосы, характеризующие метакрилатный анион, также хорошо проявляются в спектре: 2950, 2928 см-1 (валентные колебания CH связей), 1460, 1384, 1240 см-1 – скелетные деформационные колебания в узле СН3 -С=. Полосы N=C связей смещены в отличие от полимеров в более сильное поле и проявляются в области 1630-1660 см-1, причем в органоглине до полимеризации интенсивность этих полос выше, чем после полимеризации, т.е. количество привитого полимера невысокое, что подтверждается элементным анализом на азот.
Таким образом, данные ИК-спектроскопии подтверждают образование химической связи полимера с минеральным носителем.
3.4 Термофизические характеристики глины месторождения Герпегеж и синтезированных нанокомпозитов
Для изучения термофизических свойств синтезированных продуктов и исходных реагентов использовали программно-аппаратный комплекс с пакетом компьютерных программ, предназначенных для количественной обработки дериватограмм (кривых Г, TG, DTG, DTA), разработанный в Институте химии растворов РАН (г. Иваново) для измерения и регистрации выходных сигналов от датчиков дериватографа 1000D (MOM, Венгрия)
Структурная схема комплекса приведена на рис. 3.4.1. В состав комплекса входят: инструментальный усилитель с коэффициентом усиления Ку = 500, пятиканальный аналогоцифровой преобразователь (АЦП), программное обеспечение, предназначенное для работы ил IBM-совместимом компьютере с операционной системой WINDOWS 98 и выше. Выходным значением комплекса является 16-ти разрядный код АЦП. Преобразование измеряемых параметров в показание физических величин производилось с помощью функций, которые вводятся на закладке ''коэффициенты» программы ADC RQ.exe. Предполагалась жесткая привязка датчиков дериватографа к измерительным каналам АЦП:
1канал — вход датчика дифференциальной термогравиметрии (DTG),
2канал — вход датчика температуры (7),
3канал — вход датчика термогравиметрии (TG),
4канал — вход датчика дифференциального термического анализа ДТA),
5канал — вход эталонного датчика температуры.
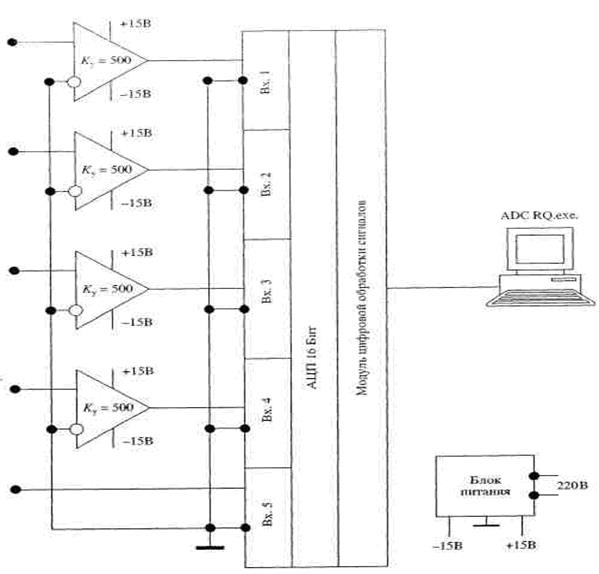
Рис. 3.4.1. Структурная схема программно-аппаратного комплекса
Объектами исследования в данной главе были:
1. образец глины Герпегеж, освобожденный от балластных веществ и модифицированный нами в натриевую форму;
2. образец «органоглины», полученной нами интеркалированием метакрилатом и акрилатом гуанидина из натриевой формы глины;
3. композиционные материалы на основе монтмориллонита и (мет)акрилата гуанидина
Анализ термогравиограммы образца натриевой глины (рис.3.4.2.)позволил выделить два участка, сопровождаемых существенным изменением массы. Уменьшение массы образца на 8% происходит при температуре до 1500С. Это связано с удалением адсорбционной воды из образца глины. И именно с потерей воды связан низкотемпературный интервал эндотермического разложения (1400С) (рис.3.4.3.). Далее, на термогравиметрической кривой в температурном интервале 150-2400С наблюдается плато, сопровождаемое незначительным падением массы (до 2%). На этом участке могут протекать следующие процессы. Во-первых, удаление структурной воды, которая в виде гидроксила входила в глинистый минерал. Во-вторых, учитывая высокое содержание железа в образце глины и карбонатных пород, можно было бы предположить наличие в образце следовых концентраций минерала сидерита FeCO3, который при температуре 350-4500С подвергается разложению и одновременным окислением оксида железа(II) до Fe2 O3 :
FeCO3 ®FeO + CO2
FeO + O2 ® Fe2 O3
Общая потеря массы составляет всего 19 %.
|
|
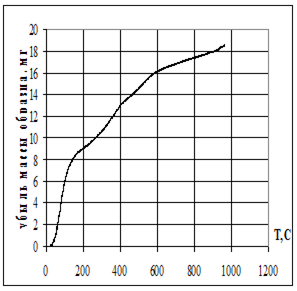
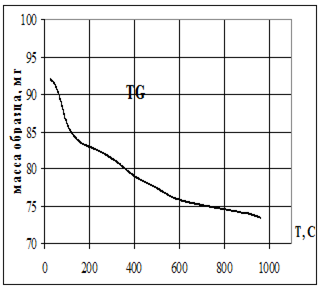
Рис.3.4.2. Термогравиограммы образца натриевой глины
|
|
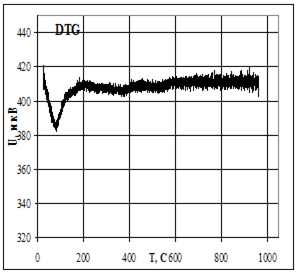
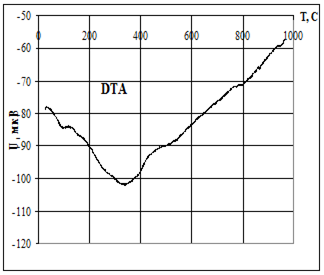
Рис.3.4.3. DTG и DTA образца натриевой глины
Процесс термического разложения органоглины более сложен. Здесь можно выделить следующие процессы:
1) удаление адсорбционной воды (до 1500С)
2) термолиз органического вещества, находящегося в межслоевом пространстве органоглины (180-4000С) с вероятным образованием соединений с высоким содержанием углерода (кокса)
3) окисление этих соединений при более высоких температурах (500-8000С)
В образце органоглины до температуры 100ºС происходит удаление воды, сопровождающееся эндоэффектом при температуре 70ºС. Вероятнее всего это адсорбционная вода. На кривой зависимости падения массы от температуры на этом участке потеря образца составляет 3, 5 %. При температуре 177ºС происходит второй эндоэффект, который не сопровождается падением массы, что вероятней всего связано с плавлением гуанидиногого производного. Далее в интервале температур 200-600ºС идет процесс термолиза гуанидинового остатка, входящего в межслоевое пространство. При этом процесс термолиза вначале сопоровождается значительным эндоэффектом при температуре 290ºС, при этом общее падение массы составляет 16 %. Уменьшение массы на 10 % наблюдается при температуре 236º С. Еще один эндоэффект наблюдается при температуре 380-390ºС, общая потеря массы при этом составляет 21 %. Далее при температуре выше 440º С начинается процесс термоокисления. Этот процесс протекает до 580ºС, общая потеря массы 34 %. Дальнейшее повышение температуры выше 600º С приводит к незначительному эндоэффекту при 700º С, который отражает дальнейшее разрушение остатков образца, общая потеря массы 42 %. Общее падение массы выше 700º С вплоть до 1000º С 47% (рис.3.4.4., 3.4.5.)
|
|
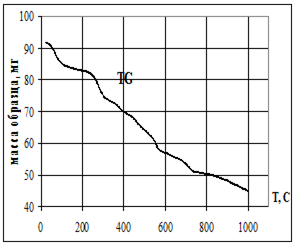
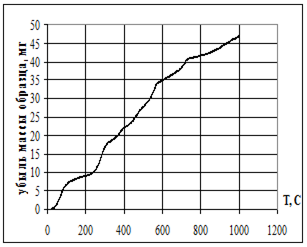
Рис.3.4.4. Термогравиограммы образца органоглины
|
|
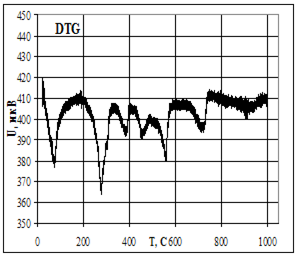
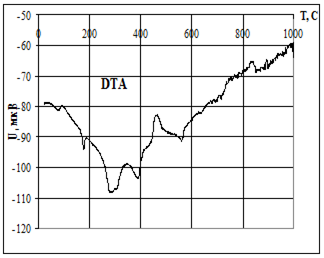
Рис.3.4.5. DTG и DTA образца органоглины
По данным работы[130] при окислении производных мочевины, в том числе и гуанидина, могут выделяться водород, угарный газ, углекислый газ, метан.
С учетом возможного термолиза гуанидина с образованием карбамида суммарная реакция термодеструкции гуанидинового остатка упрощенно может быть представлена следующей реакцией:
72СО(NH2 )2 → 45NH3 + 15CO + 15H2 O + 5N2 + 4CO2 + 17(NH2 )2 (CO)2 NH + 19NH2 CN
Анализ термических характеристик композита состава глина: МАГ 50 на 50 показал, что при температуре 100ºС имеет место сильное падение массы равное 12%. Скорее всего это связано с удалением воды с одновременным разложением самого полимера. При температурах выше 150ºС наблюдается сильный эндотермический эффект и происходит термическое разложение полимерной массы до 580º С, который сопровождается тремя термическими эффектами при 270-280º С и 430-420ºС который при температуре 500º С переходит в экзоэфект, отражающий термоокислительную деструкцию полимера. Выше 600ºС до 720º С происходит удаление коксовой массы и остается 20% твердого остатка. Процесс термолиза композит 60:40 (глина 40) соединения более сложный с большим числом стадий, отражающий термодеструкцию полимера. При температуре порядка 470ºС начинается выжигание полимера, которое заканчивается при температуре 580º С. Последняя стадия с 580 до 720º С заканчивается выгоранием кокса. Система отличается сложными процессами термической деструкции.
В образце с меньшим содержанием монтмориллонита 1:2 (глина 1) на первой стадии наблюдается уменьшение массы на 12 % при 150º С. Небольшой эндоэффект при 117º С, переходит в первый широкий эндоэффект при температуре 290º С, маленький эндоэффект при 410ºС переходит в более сильный и при температуре 480ºС превращается в экзоэффект. Общее падение массы 58% до коксообразования, при догорании кокса общая потеря массы 63 %. Чем выше концентрация полимера, тем больше остается невыгоревшего кокса в межслоевом пространстве. Характер разложения в основном определяется термической деструкцией полимерной массы находящейся в межслоевом пространстве. Основная масса вещества удаляющаяся из зоны реакции связана с летучими продуктами термолиза полимера и лишь незначительная часть вызвана процессами горения органических остатков.
Существенная разница в величине падения массы органоглины, полученной нами (общее падение массы 47%) и композиционного материала (для последнего эта величина составляет 70%) указывает на то, что доля органического реагента в композиционном материале выше, чем в образце органоглины.
3.5 Исследование сорбционных свойств композиционных материалов
Способность бентонитовых глин к поглощению различных веществ давно известна и сравнительно хорошо изучена. Глинистые минералы применяют в основном как сорбенты и коагулянты для очистки водных сред [1,2].
Основной недостаток глинистых минералов как сорбентов – это невозможность применить сорбционную технологию с их участием в колоночном варианте, т.е. динамических условиях.
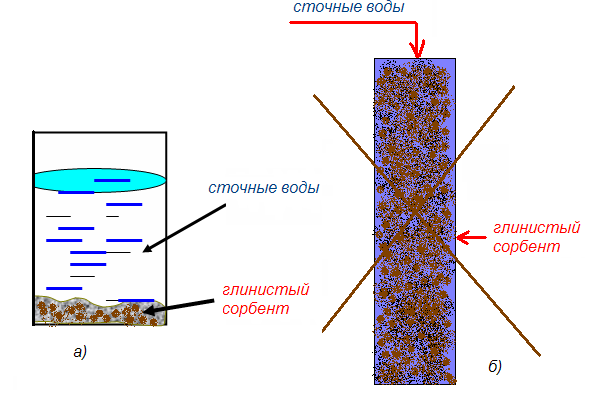
Рис. 3.5.1. Способы реализации сорбционных процессов
а) статический б) динамический
Попытка модифицировать для этого глинистые сорбенты термической обработкой при 800-12000С всегда приводит к необратимой потере сорбционных свойств глинистого минерала и понятно почему: при такой температуре происходит спекание алюмосиликатных слоев.
Выходом из этой ситуации может быть создание новых материалов на основе глинистых минералов, а именно, полимерных нанокомпозитов.
В данной работе в процессе реакции полимеризации мономера в глине, образуется композиция, которая представляет собой твердую однородную массу способную набухать в воде и обладает свойствами эффективного фильтрующего материала, в том числе и в динамических условиях очистки воды.
Ионообменные свойства композиции определяются как свойствами полимера – полиамфолита. так и катионообменными свойствами бентонитовой глины.
Исследование сорбции у полученных фаз осуществляли традиционными способами, которыми обычно пользуются для оценки «активности» сорбентов: по адсорбции метиленового синего и йода из водного раствора, в статических условиях.
Равновесную концентрацию метиленового синего (3,7-бис (диметиламино) – фетазин) после адсорбции на образцах глины и композита определяли фотометрическим методом на КФК-2 при длине волны света λ=750 нм. Зависимость адсорбции от концентрации красителя описывали эмпирическим уравнением Фрейндлиха в логарифмической форме:
![]()
где х- масса адсорбированного красителя, мг; m-масса глины, г; С- равновесная концентрация красителя мг/мл; lgа и b – константы, вычисленные МНК.
Для сравнения сорбционных возможностей исходной бентонитовой глины и композитов были проведены количественные измерения и построены кривые адсорбции.
Таблица 3.5.1.
Результаты адсорбции МС Na-формой монтмориллонита
месторождения «Герпегеж»
№. № | m масса бентонита в г | x Масса красителя сорбированного бентонитом в мг | m/x | lg(m/x) | C Равновесная концентрация красителя, мг/мл | lgC |
0.10 0.20 0.30 0.40 0.50 0.60 | 47.42 59.98 62.25 66.38 69.82 69.85 | 474.242 299.9163 207.4914 165.9587 139.6368 116.4126 | 2.676 2.477 2.317 2.22 2.145 2.066 | 0.893 0.804 0.1099 0.0500 0.00172 0.001 | -0.0493 -0.0945 -0.959 -1.301 -2.765 -3,00 |
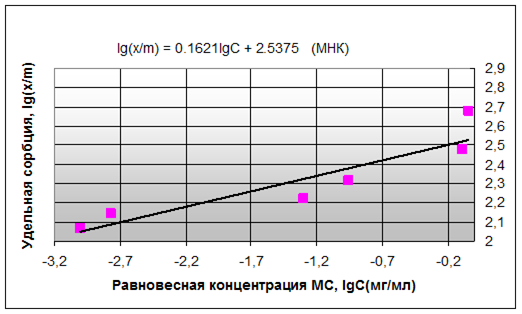
Рис.3.5.2. Изотерма адсорбции метиленового синего Na-монтмориллонитом (Герпегеж)
Таблица 3.5.2.
Результаты адсорбции МС композиционным материалом
№. № | m масса композита, в г | x Масса красителя сорбированного композитом, в мг | x /m | lg (x / m) | C Равновесная концентрация красителя, мг/мл | lgC |
0.10 0.20 0.30 0.40 0.50 0.60 0,7 0,80 0,90 | 59,0 69,76 69,9 69,91 69,92 69,93 49,88 97,3 142,0 | 590 348,8 233 174,78 139,84 116,55 249,4 486,5 711,5 | 2,771 2,541 2,367 2,242 2,145 2,066 2,396 2,686 2,855 | 1,57х10-1 3,42х10-3 1,429х10-3 1,285х10-3 1,143х10-3 1х10-3 2,4х10-3 2,7х10-2 5,133х10-2 | -0,8037 -2,465 -2,845 -2,891 -2,942 -3 -2,619 -1,568 -1,289 |
![]()
Рис. 3.5.3. Изотерма адсорбции метиленового синего композиционным материалом на основе Na-монтмориллонита и метакрилата гуанидина.
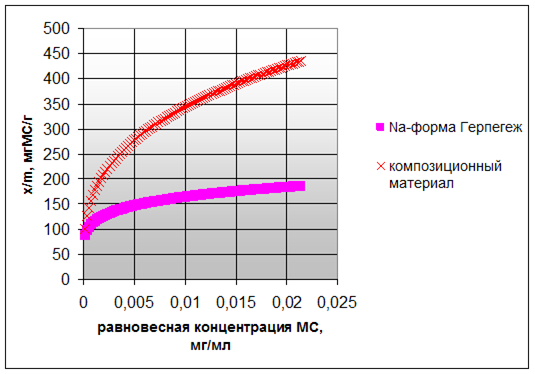
Рис.3.5.4. Сравнительная диаграмма изотерм адсорбции Na-монтмориллонита и композиционного материала ММТ+МАГ
Таблица 3.5.3.
Оценка адсорбционной емкости полимерных композитов
№/№ | образец | Адсорбционная емкость, мг*г-1 | |
по йоду | по МС | ||
1 | ММТ+МАГ (1:1) | 44 | 66 |
2 | ММТ+МАГ (1:2) | 135 | 34 |
3 | ММТ+АГ (1:1) | 165 | 36 |
4 | ММТ+АГ (1:2) | 170 | 40 |
ММТ+ММГ | 180 | 52 |
Таким образом, результаты исследований показывают о существенном увеличении адсорбционных свойств модифицированного бентонита по сравнению с исходной Na-формой глины месторождения Герпегеж.
Представляло смысл апробировать полученные сорбенты по отношению к фенолу. Фенол – один наиболее представительных органических загрязнителей, которые даже в малых количествах представляют большую опасность, как для здоровья человека, так и для окружающей среды. Попадая в воздух, воду и грунт, фенол и его производные способны вызывать генные мутации в живых организмах, а в больших количествах – влиять на видовое разнообразие. Были построены изотермы адсорбции фенола из его водных растворов при комнатной температуре.
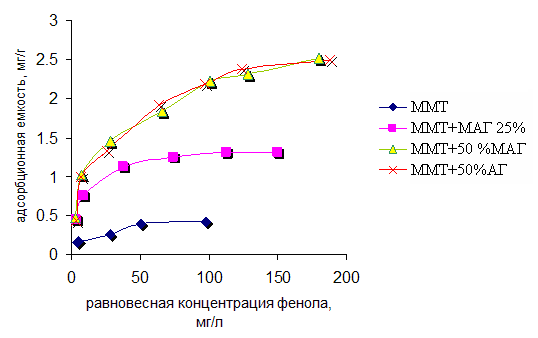
Рис.3.5.5. Изотерма сорбции фенола монтмориллонитом и гуанидинсодержащими композиционными материалами
Данные исследования показывают, что поглощательная способность сорбентов по отношению к фенолу увеличивается для композиционных материалов, которые были получены при обработке более высокой концентрацией мономеров. Интересно и то, что при содержании фенола менее 1мг/л его извлечение из раствора составляет 80-95%.
Возможность извлечения синтезированными композиционными материалами некоторых тяжелых металлов из сточных и природных вод исследовали с использованием модельных растворов.
Извлечение ионов металлов из водного раствора оценивалось следующими параметрами:
1) концентрацией ионов металла в исходном растворе и в растворе, обработанном сорбентами;
2) степенью извлечения ионов металла.
Измерения массовой концентрации металлов в пробах воды до и после обработки композитами проводили атомно-адсорбционным методом с электротермической атомизацией с использованием атомно-адсорбционного спектрометра «МГА-915».
Для исследования сорбции меди на композитах в динамических условиях раствор CuSO4 заданной концентрации в интервале 1´10-4 – 1 моль/л пропускали через колонку с предварительно набухшими в воде композитами со скоростью 0,15 мл/мин при загрузке колонки 0,1 г в пересчете на сухое вещество. Фильтрат раствора на выходе колонки собирали через фиксированные промежутки времени отдельными порциями и в каждой порции с помощью атомно-абсорбционного метода определяли концентрацию ионов меди. Результаты сорбции меди, свинца и кадмия представлены в таблице 3.5.4.
Таблица 3.5.4.
Измерения массовой концентрации металлов в пробах воды до и после обработки композитами
№ п/п | Элемент | Концентрация металла, мг/л | ||
До очистки | После очистки | Степень сорбции,% | ||
Монтмориллонит + МАГ | ||||
1 | медь | 2,91 | 0,75 | 74,22 |
2 | свинец | 1,38 | 0,284 | 79,42 |
3 | кадмий | 1,84 | 0,309 | 83,20 |
Монтмориллонит + АГ | ||||
1 | медь | 2,91 | 0,047 | 98,3 |
2 | свинец | 1,38 | 0,192 | 86,08 |
3 | кадмий | 1,84 | 0, 205 | 88,85 |
Как видно из таблицы, синтезированные гуанидинсодержащие композиты проявляют достаточно высокую сорбционную активность в отношении изученных металлов, причем у композита с полиакрилатом гуанидина наблюдается наиболее выраженная способность связываться с ионами металлов.
3.6 Исследование степени набухаемости, плотности и удельной поверхности монтмориллонита и композиционных материалов на его основе
Под набухаемостью понимают способность глинистых пород увеличивать объем в процессе взаимодействия с водой или водными растворами [110]. Процесс набухания сопровождается увеличением влажности, объема породы и возникновением давления набухания.
Процесс набухания проходит в две стадии: первая стадия — адсорбционное или внутрикристаллическое набухание, вторая — макроскопическое или «осмотическое» набухание. На первой стадии глинистая порода впитывает влагу за счет адсорбции молекул воды поверхностью глинистых частиц и межслоевыми промежутками кристаллической решетки глинистых минералов. Эта стадия практически не влияет на изменение объема породы. На второй стадии набухания поглощение влаги осуществляется с помощью осмотического давления. Оно возникает вблизи поверхности глинистых частиц за счет избыточной концентрации многочисленных обменных катионов отдиссоциированных (отошедших) с поверхности глинистых частиц в раствор. Основное увеличение объема набухающей глины происходит именно на этой макроскопической стадии.
Способность глин к набуханию характеризуется влажностью набухания (Wн ) и давлением набухания (Pн ). По величине давления набухания глинистые породы подразделяются на ненабухающие (Pн < 0,025 МПа); слабо набухающие (Pн = 0,025 – 0,1 МПа); средненабухающие (Pн = 0,1 – 0,25 МПа) и сильнонабухающие (Pн > 0,25 МПа) [131]. Величина и характер набухания глинистых пород определяются многими факторами, основными из которых являются минеральный состав, дисперсность и структура. Наибольшим набуханием обладают глинистые породы, в составе которых имеются глинистые минералы с подвижной кристаллической структурой (например, монтмориллонит), наименьшим — минералы с более жесткой кристаллической структурой (каолинит). Сильное влияние на набухание глин оказывает и их структура, при этом определяющее значение имеет характер структурных связей и тип контактов между минеральными частицами. Наибольшее набухание характерно для глинистых пород с переходными контактами, наименьшее — для глин с фазовыми контактами.
Глинистые породы, обладающие преимущественной ориентацией структурных элементов, характеризуются ярко выраженной анизотропией набухания. Наибольшее набухание отмечается в направлении, перпендикулярном ориентации частиц. В ходе процесса набухания происходит существенная перестройка исходной микроструктуры глинистой породы. Пористость набухших глин в конце процесса набухания может достигать 85-90%.
Результаты измерения плотности и степени набухаемости исходного и модифицированного монтмориллонита в воде приведены в таблице 3.6.1., 3.6.2.
Таблица 3.6.1.
Определение степени набухания композитов в воде
№ | Образец | Плотность, r, г/см3 | m1 сухого образца, г | m2 набухшего образца, г | Степень набухания F |
1 | Монтмориллонит Ca | 4,0574800 | 0,2 | 0,6 | 3,0 |
2 | Монтмориллонит Na | 1,5503832 | 0,2 | 1,2 | 6,0 |
3 | органоглина | 8,17143219 | 0,2 | 0,8 | 4,0 |
4 | ММТ+ АГ | 1,4522410 | 0,2 | 1,0 | 5,0 |
5 | ММТ + МАГ | 1,5631753 | 0,2 | 1,4 | 7,0 |
Таблица 3.6.2.
Определение степени набухания композитов в мочевине и фурациллине
№ | Образец | Плотность, r, г/см3 | Степень набухания F в мочевине | Степень набухания F в фурациллине |
1 | монтмориллонит Ca | 4,0574800 | 3,2 | 3,5 |
2 | Монтмориллонит Na | 1,5503832 | 7,0 | 18 |
3 | органоглина | 8,17143219 | 10 | 8,75 |
3 | ММТ+ АГ | 1,4522410 | 5 | 4,5 |
4 | ММТ + МАГ | 1,5631753 | 3,5 | 3,2 |
Как видно из полученных данных, синтезированные гуанидинсодержащие композиты проявляют достаточно высокую степень набухаемости в воде и в водных растворах органических веществ, причем в органических веществах и монтмориллонит и органомодифицированные глины характеризуются большей степенью набухаемости. Исходя из наличия в молекулах фурациллина и мочевины групп CO и NH можно ожидать их высокую способность к образованию водородных связей. По-видимому, эти органические соединения разрушают водородные связи между кристаллитами бентонита и внедряются между ними за счет образования новых связей.
Удельную поверхность и распределение частиц по размерам определяли с использованием лазерного анализатора частиц. Для композита на основе Ca-монтмориллонита и МАГ Sуд. =7,39´102 см2 /г (диаметр частиц меньше 0,76), Na- монтмориллонита+МАГ Sуд. =9,62´102 см2 /г, Na- монтмориллонита+АГ Sуд. =6,23´102 см2 /г. Как видно, композиционные материалы имеют достаточно высокую удельную поверхность частиц, что подтверждает возможность их использования в качестве сорбентов.
Тем не менее, наиболее активный образец Na- монтмориллонит+АГ (табл. 3.6.1., 3.6.2.) имеет меньшую площадь удельной поверхности и большую долю крупных частиц, чем другие образцы. Такое отсутствие простой зависимости размера гранул от площади поверхности свидетельствует, видимо, о большей роли микроструктуры кристаллов, их поверхностной неоднородности. В работе [132] отмечается, что однозначная связь между размерами гранул, площадью поверхности и реакционной способностью образцов наблюдается лишь при их незначительной пористости. В случае же большого количества дефектов поверхность приобретает фрактальные свойства [133]. Так, расчет авторов работы [132] для некоторых, особенно дефектных, образцов биогенного карбоната кальция показал, что так называемая «реакционная площадь поверхности» составляет лишь 10—30% от величины, определенной методом БЭТ. Очевидно, композиционные материалы, полученные с использованием в качестве органомодификатора акрилата гуанидина, имеют более дефектную структуру и как следствие, в значительной степени обладает фрактальными свойствами.
Таким образом, исследование удельной поверхности и распределения частиц по размерам образцов разного состава, показало отсутствие простой зависимости размера гранул от площади поверхности, определенной лазерным анализатором частиц, что свидетельствует о значительной роли микроструктуры кристаллов и высокой пористости полученных нанокомпозитов
3.7 Биоцидные свойства гуанидинсодержащих полимерных композиционных материалов
Предварительные исследования бактерицидной активности синтезированных композиционных материалов, проведенные совместно с Бактериологической лабораторией ГСЭН КБР, показали, что они весьма активны и обладают биоцидным действием по отношению к некоторым микробиологическим загрязнителям воды, в частности к кишечной палочке.
Для определения числа колоний кишечной палочки производили посев исследуемой воды, обработанной гуанидиновыми композитами, на мясо-пептонную среду и выращивали колонии E.coli в течение 48 часов при 22ºС, после чего колонии пересчитывали.
Эффективность бактерицидного действия композитов определяли по количеству колониеобразующих единиц (КОЕ), рассчитывая их жизнеспособность по формуле
С = lg Nt /Ntk ,
где Nt – количество бактерий, вышивших после обработки композитом,
Ntk — количество бактерий в контроле за один и тот же промежуток времени.
Выявлено, что синтезированные композиты характеризуются 70,0 (композит с МАГ) и 85,0 % (композит с АГ) бактерицидной активностью по отношению к кишечной палочке. Примечательно, что исходные мономеры и соответствующие гомополимеры проявляют по отношению к кишечной палочке недостаточно высокую биоцидную активность. Видимо, в данном случае вклад в биоцидный эффект вносит и монтмориллонит.
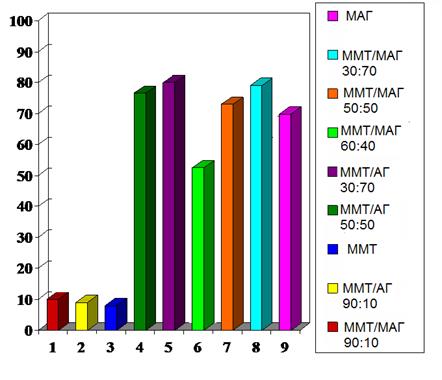
Рис. 3.7.1. Сравнительная биоцидная активность гуанидинсодержащих композиционных материалов по отношению к Е-coli
Таким образом, сочетание в полученных материалах высокой бактерицидной активности с повышенной способностью связываться с тяжелыми металлами и органическими поллютантами открывает возможности их использования в качестве эффективных сорбентов для очистки и обеззараживания воды.
ВЫВОДЫ
1. Радикальной полимеризацией в растворе с использованием в качестве мономеров акрилата и метакрилата гуанидина, органомодифицированного слоистого силиката в качестве наномерного наполнителя впервые синтезированы полимерные гуанидинсодержащие нанокомпозиты.
2. Совокупностью физико-химических методов исследования (ИК-спектроскопия, ренгеноструктурный анализ, дифференциальный термический анализ, лазерный анализатор частиц) изучены состав и структура синтезированных композитов.
3. Оценены сорбционные свойства новых гуанидинсодержащих композиционных материалов с использованием статического и динамического режима очистки воды
4. Обнаружено, что синтезированные композиты обладают высокой сорбционной способностью к ионам тяжелых металлов и органическим загрязнителям воды
5. Изучены бактерицидные свойства нанокомпозитов и показано, что они обладают значительной биоцидной активностью по отношению к грампополжительным и грамотрицательным микроорганизмам
6. Впервые получены эффективные сорбенты на основе монтмориллонита и гуанидинсодержащих полимеров, обладающие высокими сорбционными и антимикробными характеристиками
ЛИТЕРАТУРА
1. М.А. Микитаев, О.Б. Леднев, А.А. Каладжян, Б.З. Бештоев, А.Ю. Беданоков, А.К. Микитаев Полимерные нанокомпозиты на основе органомодифицированных слоистых силикатов – новый тип конструкционных материалов // II Международная конференция «Новые полимерные композиционные материалы», июль 2005г.
2.Микитаев А.К., Каладжян А.А., Леднев О.Б., Микитаев М.А., Давыдов Э.М… Нанокомпозитные полимерные материалы на основе органоглин с повышенной огнестойкостью // Пластич.массы. – 2005. – №4. – C. 26-31.
3. Kojima Y., Usuki A., Kawasumi M., Okada A., Kurauchi Т., Kamigaito О. Synthesis of nylon 6-clay hybrid by montmorillonite intercalated with caprolactam.
J. Polym. Sci., Part A, 1993, v. 31, p .983-986.
4. Kojima Y., Usuki A., Kawasumi M., Okada A., Kurauchi Т., Kamigaito O. One-pot synthesis of nylon 6-clay hybrid. J. Polym. Sci., Part A, 1993, v. 31, p. 1755-1758.
5. K. Yano, A. Usuki, A. Okada, T. Kurauchi, O. Kamigaito. Synthesis and properties of polyimide-clay hybrid, J. Polym. Sci.: Part A: Polym. Chem. 31 1993, p. 2493-2498
6. Костандов Л. A., Ениколопов Н. С., Дьячковский Ф. А., Новокшонова Л. А., Кудинова О. И., Маклакова Т. А., Гаврилов Ю. А., Акопян Л. А., Брикенштейн Х-М. А. Авт. Свид. СССР 763379 (1976); Бюл. изобр.,1980, №34, с. 129.
7. Ломакин С.М., Заиков Г.Е. Высокомолек. Соед. Б.2005.-Т47.№1- С.104-120.
8. Колесников Ю. Н., Холстов Г. П., Шишлов С. С, Махинько А. И., Шевцова Л. С., Дьячковский Ф. А., Белов Г. П., Новокшонова Л. А., Корнеев Н. Н., Казаков Ю. М., Соловьёва Т. И., Гоменюк В. Я. В сб.: комплексные металлоорганические катализаторы полимеризации олефинов. Черноголовка: Изд. ИФХ АН СССР, 1982. №9, с. 30
9. Alexandre М., Dubois Ph. Polymer layered silicate nanocomposites: preparation,properties and uses of a new class of materials. Mater. Sci. and Eng.,
2000, V. 28, p. 1-63.
10. Ray S. S., Okamoto M. Polymer/layered silicate nanocomposites: a review from preparation to processing. Prog. Polym. Sci., 2003, v. 28, p. 1539-1641.
11. А.Х. Маламатов, Г.В. Козлов, М.А. Микитаев. Механизмы упрочнения полимерных нанокомпозитов. Москва. РХТУ. 2006 г. -239 с.
12. Нильсен Л. Е. Механические свойства полимеров и полимерных композиций. / Пер. с англ. канд. техн. наук П. Г. Бабаевского. — М.: «Химия»,1978. -312 с.
13. Липатов Ю. С. Физическая химия наполненных полимеров; М.: «Химия»,
1977. 304 с.
14. Gonsalves К.Е., Chen X. «Inorganic nanostructured materials». Nanostructured materials, 1996, v. 5, p. 3256.
15. Материаловедение: Учебник для вузов. / Б.Н. Арзамасов, В.И. Макарова
и др. Под общ. ред. Б.Н. Арзамасова, Г.Г. Мухина. — 3-е изд., переработ, и доп. — М.: Изд-во МГТУ им. Н.Э. Баумана, 2001. – 648 с. Ил.135
16. Mark J.E. Ceramic reinforced polymers and polymer-modified ceramics. Polym. Eng. Sci. — 1996. — № 36. — P. 2905-2920. Reynaud E., Gauthier C, Perez J. Nanophases in polymers. Rev. Metall. Cah. Inf. Tech. — 1999. — № 96. P. 169-176
17. Werae Т., Patten Т.Е. Preparation of structurally well defined polymernanoparticle hybrids with controlled/living radical polymerization. J. Am,
Chem. Soc. — 1999. -№121. — P. 7409-7410.
18. Herron N., Thorn D.L. Nanoparticles. Uses and relationships to molecular
clusters. Adv. Mater. — 1998. — №10. — P. 1173-1184.
19. Calvert P. Potential applications of nanotubes, in: T.W. Ebbesen (Ed.), Carbon Nanotubes. CRC Press. Boca Raton. FL. — 1997. — P. 277-292.
20. Favier V., Canova G.R., Shrivastava S.C, Cavaille J.Y. Mechanical percolation
in cellulose whiskers nanocomposites. Polym. Eng. Sci. — 1997. -№37 — P. 1732-1739.
21. Chazeau L., Cavaille J.Y., Canova G., Dendievel R., Boutherin B. Viscoelastic
properties of plasticized PVC reinforced with cellulose whiskers. // J.Appl. Polym. Sci. — 1999. — №71. — P. 1797-1808.
22. Theng B.K. The Chemistry of Clay-Organic Reactions. — New York: Wiley, 1974.
23. Ogawa M., Kuroda K. Preparation of inorganic-organic nanocomposites through intercalation of organoammonium ions into layered silicates. // Bull. Chem. Soc. Jpn.- 1997. -№70, p. 2593-2618.
24. Kryszewski М. Nanointercalates — novel class of materials with promising properties. // Synthetic Metals. — 2000, №109. p. 47-54.
25. Браун Г. Рентгеновские методы изучения и структура глинистых минералов. М., «Мир», 1965, 600 с.
26. Theng В. К. Formation and properties of clay-mineral complexes. Amsterdam, Elsevier, 1979, 112.
27. Грим Р.Е. Минералогия глин. М.: Изд. иностр. лит. 1959.- 452 с.
28.Куковский Е.Г. Особенности строения и физико-химические свойства глинистых минералов, М.: Химия, 1966г, 158с.
29.Карпова Г.В. Глинистые минералы и эволюция в терригенновых отложениях. М., Недра, 1978, с.408с.
30.Горбунов Н.И. Высокодисперсные минералы и методы их изучения Москва: Госхимиздат, 1963г, 402с.
31.Кристаллохимия и спектроскопия минералов Киев: «Наукова думка», 1984г., с. 437.
32.Злочевская Р.И. Связанная вода в глиняных грунтах М.: МГУ, 1969, 198с.
33.Круглицкий И.И. Физико-химические основы регулирования свойств дисперсий глинистых минералов, Киев, 1968г., 311с.
34.Куколев Г.В. Химия кремния и физическая химия силикатов. М.: Химия, 1985г, 291с.
35.Мюллер А. Химическая петрология, М.: Мир, 1980, 387с.
36.Молекулярная спектроскопия и рентгенография минералов Л.: «Недра», 1985г, 422с.
37.Вертушков Г.Н. Таблицы для определения минералов по физическим и химическим свойствам Л.: Недра, 1984г., 320 с.
38.Термический анализ минералов и горных пород, Л.: Недра, 1974, 412 с.
39. Maegdefrau E., Hofmann U. Die Kristallstruktur des Montmorillonits. // Z.
Krist. 1937, №98, p. 299-323.
40. Marshal С E. Layer Lattices and base-exchange clays. // Z. Krist, 1935, №91, p. 433-449.
41. Рентгенография основных типов породообразующих минералов. Под ред. В. А. Франк-Каменецкого. Л.: «Недра», 1983.
42. Фридрихсберг Д.А., Курс коллоидной химии. Л: «Химия», 1974, 350 с.
43. Жукова А.И., Вдовенко Н.В., Калашникова Л.Е. Ионообменное взаимодействие четвертичных алкиламмониевых катионов с Na- Са-формами монтмориллонита — Укр. хим. журн., 1975, т. 41, № 7, с. 696 -699.
44. Морару В. Н., Маркова С. А., Овчаренко Ф. Д. Адсорбция катионных поверхностно-активных веществ на монтмориллоните из водных растворов.
Укр. хим. журн., 1981, т. 47, № 10, с. 1058-1064.
45. Ширинская Л.П., Ермоленко Н.Ф. Сорбция органических катионов на замещённых формах глин. Коллоид, журн., 1962, т. 21, № 3, С. 340 — 343.
46. Yang J.-H., Han Y.-S., Choy J.-H., Tateyama H., — J. of Mater. Chem., 2001,V.11,p. 1305.
47. Герасин В. A., Гусева М. A., Бахов Ф. Н., Каргина О. В., Мерекалова Н. Д., Антипов Е. М., «Формирование, структура и свойства органофильных слоев на Na+-MMT, образованных модификатором ДОДАБ», Научный семинар" Актуальные проблемы реологии, 30 июня-5июля 2003г., Барнаул.
48. Vaia R. А., Teukolsky R. К., Giannelis Е. Р., Interlayer structure and molecular environment of alkylammonium layered silicates,Ghem. Mater, 1994, V.6, p. 1017-1026.
49. Герасин В. A., Бахов Ф. Н, Королёв Ю. М., Мерекалова Н. Д.,. Fischer Н. R., Антипов Е. М. / Структура формирующихся на Na-монтмориллоните слоев поверхностно — активных веществ и совместимость модифицированной глины с полиолефинами. / Высокомолек. соед., 2005,№ 8, в печати.
50. Vaia R.A., Jandt K.D., Kramer E.J., Giannelis E.P., Kinetics of polymer melt intercalation, Macromolecules 28, 1995, p. 8080-8085.
51. B.A. Герасин, M. A. Гусева, A.A. Баранников, Б.Ф. Шклярук, Ю.М. Королёв, «Нанокомпозиты на основе полиолефинов и природной глины». Всероссийская конференция с международным участием «Современные проблемы химии высокомолекулярных соединений: высокоэффективные и экологически безопасные процессы синтеза природных и синтетических полимеров и материалов на их основе», 20-27 августа 2002г., Улан-Удэ, с. 49.
52. Fermeglia М., М. Ferrone, S. Pricl, Computer simulation of nylon-6/organoclay nanocomposites: prediction of the binding energy. Fluid Phase Equlilibria, 2003, v. 212, p. 315-329.
53. V. Kuppa, T. M. D. Foley, E. Manias, Segmental dynamics of polymers in nanoscopic confinements, as probel by simulations of polymer/layeredsilicate nanocomposites. Eur. Phys. J. E 12, 2003, p. 159-165.
54. W. Xiao, M. Zhan, Z. Li, Organically modifying and modeling analysis of montmorillonites. Materials and Design 24, 2003, p. 455-462.
55. Помогайло A. Д., Розенберг A. С, Уфлянд И. Е. Наночастицы металлов в полимерах. Москва, «Химия», 2000, 672 с, М. «Химия», 671 с.
56. Vaia R.A., Giannelis Е.Р. Lattice model of polymer melt intercalation in organically-modified layered silicates. Macromolecules 1997, v.30, p.7990 — 8000.
57. Vaia R.A., Giannelis E.P. Polymer melt intercalation in organicallymodified
layered silicates: model predictions and experiment. Macromolecules 1997, V. 30, p. 8000-8009.
58. Balazs A.C., Singh Ch., Zhulina E. Modeling the interactions between polymers and clay surfaces through self- consistent field theory. Macromolecules
1998, V. 31, p. 8370 — 8382.
59. Balazs A.C., Singh Ch., Zhulina E., Lyatskaya Yu. Phase behavior of polymer-clay nanocomposites. Accounts of chemical research, 1999, v. 32, p. 651-663.
60. Ginzburg V.V., Singh Ch., Balazs A.C. Theoretical phase diagrams of polymer-clay composites: the role of grafted organic modifiers. Macromolecules
2000, V. 33, p. 1089 — 1101.
61. Jeon H.G., Jung H.-T., Lee S.W., Hudson S.D., Моrfo1оgу of polymer silicate nanocomposites. High density polyethylene and a nitrile, Polym. Bull. V.41, 1998, p. 107-113.
62. Ogata N., Jimenez G., Kawai H,, Ogihara Т., Structure and thermal/mechanical properties of poly(L-lactide)-clay blend, J. Polym. Sci. Part B: Polym. Phys. v. 35, 1997, p, 389-396.
63. Jimenez G, Ogata N., Kawai H., Ogihara Т., Structure and themal/mechanical properties of poly(ε-caprolactone)- clay blend, J. Appl. Polym. Sci. V. 64, 1997, p. 2211-2220.
64. R. Vaia R. A., Giannelis E. P., Polymer melt intercalation in organically -modified layered silicates: model predictions and experiment. Macromolecules
V.30, 1997, p. 8000-8009.
65. HasegawaN., Kawasumi M;, Kato M., Usuki A., Okada A., Preparation and mechanical properties of polypropylene-clay hybrids using a maleic anhydride- modified polypropylene oligomer, J. Appl. Polym. Sci. v. 67, 1998, p. 87-92.
66. Manias E., Touny A., Wu L., Strawhecker K., Lu В., and Chung T. G., Polypropylene/Montmorillonite Nanocomposites. Review fthe Synthetic Routes and Materials Properties,Chem. Mater., v. 13, 2001, p. 3516-3523.
67. Mehta Sameer, Mirabella Francis M., Rufener Karl, Bafna Ayush Thermoplastic Olefin/Clay Nanocomposites: Moфhologyand Mechanical Properties, J. Appl.Polymer Science, v. 92, 2004, p. 928-936.
68. Kaempfer D., Thomann R., Mulhaupt R., Melt compaounding of polypropylene nanocomposities containing organophilic layered silicates and in situ formed core/shellnanoparticles. Polymer, v. 43,2002, p. 2909-2916.
69. Morgana A. B, Harrisb J. D., Effects of organoclay Sox let extraction on mechanical properties, flammability properties and organoclay dispersion of polypropylene nanocomposites. Polymer, v. 44, 2003, p. 2313-2320.
70. Hasegawa Naoki, Usuki Arimitsu, Silicate Layer Exfoliation in Polyolefin/ Clay Nanocomposites Based on Maleic Anhydride Modified Polyolefins and Organophilic Clay, J. Applied Polymer Science, Vol. 93, 2004, p. 464-470.
71. Xu W., Liang G., Zhai H., Tang S., Hang G., Pan W.P., Preparation and crystallization behaviour of PP/PP-g-MAH/Org-MMT nanocomposite, European
Polymer Journal, v.39, 2003, p. 1467-1474.
72. Danumah C, Bousmina M., Kaliaguine S., Novel Polymer Nanocomposites
from Templated Mesostructured Inorganic Materials, Macromolecules, v.36, 2003, p. 8208-8209.
73. Wang Z. M., Nakajima H., Manias E., Chung T. C., Exfoliated PP/Clay Nanocomposites Using Ammonium-Terminated PP as the Organic Modification
for Montmorillonite. Macromolecules, v. 36, 2003, p.8919-8922.
74. Greenland D.J., Adsoption of polyvinylalcohols by montmorillonite, J. Colloid Sci., v. 18, 1963, p. 647-664.
75. Ogata N., Kawakage S., Ogihara Т., Poly(vinyl alcohol)-clay blend prepared using water as solvent, J. Appl. Polym. Sci., v. 66, 1997, p. 573-581.
76. Parfitt R.L., Greenland D.J., Adsoption of poly (ethylene glycols) on montmorillonites, Clay Mineral, v.8. 1970. p. 305-323.
77. Zhao X., Urano K., Ogasawara S., Adsoption of polyethylene glycol from aqueous solutions on montmorillonite clays. Colloid Polym. Sci., v. 67, 1989, p. 899-906.
78. Ruiz-Hitzky E., Aranda P., Casal В., Galvan J.C., Nanocomposite materials with controlled ion mobility, Adv.Mater., v. 7, 1995, p. 601 — 620.
79. Billingham J., Breen C, Yarwood J., Adsorption of polyamine, polyacrylic acid and polyethylene glycol on montmorillonite: an in situ study using artier, Vibr. Spectrosc, v. 14, 1997, p. 19-34.
80. Levy R., Francis C.W., Interlayer adsorption of polyvinylpyrrolidone on montmorillonite, J. Colloid Interface Sci., v. 50, 1975, p. 442-450.
81. J. Wu, M.M. Lemer, Structural, thermal, and electrical characterization of layered nanocomposites derived from sodium-montmorillonite and polyethers, Chem. Mater., v. 5, 1993, p. 835-838.
82. Lee D.C., Jang L.W., Preparation and characterization of PMMA-clay hybrid
composite by emulsion polymerization, J. Appl. Polym. Sci., v. 61, 1996, p. 1117-1122.
83. Noh M.W., Lee D.C., Synthesis and characterization of PS-clay nanocomposite
by emulsion polymerization, Polym. Bull., v. 42, 1999, p, 619-626.
84. Lan Т, Kaviratna P.D., Pinnavaia T.J., On the nature of polyimide-clay hybrid composites, Chem. Mater., v. 6, 1994, p. 573-575.
85. Fukushima Y., Okada A., Kawasumi M., Kurauchi Т., Kamigaito O., Swelling behavior of montmorillonite by poly-6-amide, Clay Mineral, v. 23, 1988, p, 27-34.
86. Usuki A., Kojima Y., Kawasumi M., Okada A., Fukushima Y., Kurauchi Т., Kamigaito O., Synthesis of nylon-6-clay hybrid, J. Mater, Res,, v. 8, 1993, p. 1179-1183.
87. Messersmith P.B., Giannelis E.P., Synthesis and barrier properties of poly(e-caprolactone)-layered silicate nanocomposites, J. Polym. Sci.: Part A Polym. Chem, V. 33, 1995, p. 1047-1057.
88. Zilg C, MuElhaupt R., Pinter J., Morphology and toughness/stiffness balance of nanocomposites based upon anhydride-cured epoxy resins and layered silicates, Macromol. Chem, Phys., v, 200, 1999, p. 661-670.
89. Tudor J., Willington L., O'Hare D., Royan B., Chem. Commun,, 1996, p.2031-2032.
90. Heinemann J., Reichert P., Thomann R., Mulhaupt R., Macromol.Rapid
Commun., v. 20, 1999, p. 423-430.
91. Alexandre M., Dubois P., Sun T., M J., Jerome Garces, Polyethylene — layered silicate nanocomposites prepared by the polymerization — filling techiq: synthesis and mechanical properties, Polymer, v, 43,2002, p. 2123-2132.
92. Shin S.-Y. A., Simona L. C, Soaresa J. B.P., Scholzb G., Polyethyleneclay hybrid nanocomposites: in situ polymerization using bifunctional organic modifiers, Polymer, v. 44, 2003, p. 5317-5321.
93. Weil., Tang T., Huang B., Synthesis and characterization of polyethylene/ clay-silica nanocomposites: montmorillonite/silica-hybrid-supported catalyst and in situ polymerization, J. of Polymer Science. Part A: Polymer Chemistry, v. 42, 2004, p. 941-949.
94. Liu C, Tang Т., Wang D., Huang В., In situ ethylene homopolymerization and copolymerization catalyzed by zirconocene catalysts entrapped inside functionalized montmorillonite, J Polym Sci., Part A., Polym Chem. v. 41,2003, p. 2187-2196.
95. Yang F., Zhang X., Zhao H., Chen В., Huang В., Feng Z., Preparation and properties of polyethene/montmorillonite nanocomposites by in situ polymerization, J Appl. Polym. Sciece, v. 89,2003, p. 3680-3684.
96. Harris D.J., Bonagamba T.J., Schmidt-Rohr K., Conformation of poly(ethyIene oxide) intercalated in clay and MoS2 studied by twodimensional double-quantum NMR, Macromolecules, v. 32, 1999, p. 6718-6724.
97. Vaia R.A., Vasudevan S., Krawiec W., Scanlon L.G., Giannelis E.P., New polymer electrolyte nanocomposites: melt intercalation of poly(ethylene oxide)\in mica-type silicates. Adv. Mater., v. 7, 1995, p. 154-156.
98. Dennis Н. R., Hunter D. L., Chang D., Kim S., White J. L., Cho J. W., Paul D. R., Effect of melt processing conditions on the exfoliation in organoclay- based nanocomposites, Polymer., v. 42, 2001, p. 9513-9522.
99. Morgana A. В., Harris J. D., Exfoliated polystyrene-clay nanocomposites synthesized by solvent blending with sonication. Polymer, v.45, 2004, p.8695-8703
100. Hudson S. D. «Polyolefin nanocomposites.» United States patent 5,910,523 1999.
101. Kojima Y., Usuki A., Kawasumi M., Okada A., Fukushima Y., Karauchi Т., Kamigaito O. Mechanical properties of nylon-6/clay hybrid., J. Mater. Res. -1993, №6, p. 1185-1189.
102. Liu L.M., Qi Z.N., Zhu X.G. Studies on nylon-6 clay nanocomposites bymelt-intercalation process. J. Appl. Polym. Sci., 1999, №71, p. 1133-1138.
103. Lagaly G., Smectic clays as ionic macromolecules, in: A.D. Wilson, H.J. Prosser (Eds.), Development in Ionic Polymers, Elsevier, London, 1986, pp. 77-140.
104. Черепанов Г.П. Механика хрупкого разрушения. — М. «Наука», 1974.
105. Yang Y., Zhu Z.-K., Yin J., Wang X.-Y., Preparation and properties of hybrids of organo-soluble polyimide and montmoriUonite with various chemical surface modifications methods. Polymer, 1999, №40, p. 4407-4414;
106. Bazhenov S.L. Stable crack growth in ductile polymers. J. Mater. Sci., 1997. v. 32, p. 797-802.
107. Yano К., Usuki A., Okada A., Synthesis and properties of polyimide-clay hybrid films, J. Polym. Sci., A: Polym. Chem., v. 35, 1997, p, 2289-2294.
108. Scherer C, PA Film grade with improved barrier properties for flexible food packaging applications, in: Proceedings of the New plastics'99, London, 2-4 February 1999, p. 56.
109. Tortora M., Gorrasia G., Vittoriaa V., Gallib G., Ritrovatib S., Chiellinib E., Structural characterization and transport properties of organically modified montmorillonite/polyurethane nanocomposites. Polymer, v. 43,2002, p. 6147-6157.
110.Евсикова О.В., Стародубцев С.Г., Хохлов А.Р. Синтез, набухание и адсорбционные свойства композитов на основе полиакриламидного геля и бентонита натрия. Высокомолек. Соед. Сер.А.2002. Т.44.№5 с.802-808.
111.Houben-Weil. V.8, p.180.
112.Патент Франции 789429 (1959).
113.Патент США 2,867,562 (1959).
114.Патент Великобритании 1114155 (1960).
115.Патент Швеции 339076 (1971).
116.А.с. СССР 847893 (1981).
117.Патент Великобритании 115243 (1969).
118.Патент ФРГ 2437844 (1982).
119.Патент США 4,587,266 (1986).
120.А.с. СССР 4341826 (1988).
121.Гембицкий П.А., Лиманов В.Е. //Журнал прикладной химии. 1975, №48, с.1833.
122.А.с. СССР 1616898 (1990).
123.Khokhlov A.R., Pavlova S.A., Timofeeva G.L. // J. Polymer. 1994, V.35, №8, p. 1769.
124.А.с. СССР 2039735 (1995).
125.Приказ Минздрава СССР № 15-6/31 от 22 декабря 1989 года.
126.Временное наставление по применению метацида, полисепта и фогуцида для ветеринарной дезинфекции № 22-157 от 26.12. 1991г.
127. Хаширова С.Ю. Дисс. канд. хим. наук. М. РХТУ. 2002 г.
128.К.Е. Скворцова, Нехорошева А.Г., Гембицкий П.А. // Проблемы дезинфекции и стерилизации. М.: ВНИИДиС, 1974, вып.23, с.58.
129. Сивов Н.А., Малкандуев Ю.А., Хаширова С.Ю. и др. Состав и строение сополимеров на основе акрилат и метакрилатгуанидинов // Известия Вузов. Северо-Кавказский регион. Сер. Естественные науки. Приложение № 4. 2006 С.53-65
130. К. Ингольд., Успехи химии,33,1107,1964
131.Грунтоведение / Под ред. Е.М.Сергеева. М.: Изд-во МГУ, 1983.389 с.
132. Walter L.M., Morse G.M. J. Sediment. Petrol. 1984. V.54. N4. P.1081-1090.
133. Avnir D., Pann D., Pfeifer P. // N a t u r e. 1984. V.308. N 15. P. 261-263.