Реферат: Радикальная сополимеризация акрилат- и метакрилатгуанидинов с виниловыми мономерами
Радикальная сополимеризация акрилат- и метакрилатгуанидинов с виниловыми мономерами
Введение
Развитие науки и техники выдвигает на современном этапе проблемы получения новых полимерных материалов с заданным комплексом свойств. Именно поэтому в последние десятилетия в области химии высокомолекулярных соединений интенсивное развитие получило создание и исследование синтетических полиэлектролитов. Они находят широкое применение в самых разных областях промышленности, техники, сельского хозяйства и медицины, и в дальнейшем их роль и значение, несомненно, будут возрастать.
Особый интерес представляет синтез новых сополимеров различного состава на основе акрилат- и метакрилатгуанидинов. Известно, что соединения, содержащие в своем составе гуанидиновую группу, обладают широким спектром бактерицидного действия и нередко используются в качестве лечебных препаратов, бактерицидов и фунгицидов. Введение гуанидиновой группы в полимерные продукты должно придавать им значительную биоцидную активность. Это особенно актуально для водных растворов флоккулянтов, в частности, полиакриламида, который в присутствии бактерий и плесени легко подвергается микробиологической деструкции.
При радикальной полимеризации и сополимеризации водорастворимых мономеров природа реакционной среды существенно влияет на кинетические параметры синтеза и характеристики образующихся сополимеров. Это обусловлено изменением реакционной способности реагирующих частиц вследствие их ионизации, сольватации, комплексообразования и межмолекулярных взаимодействий в реакционной среде. Поэтому осложнённый характер сополимеризации ионогенных мономеров также определяет актуальность изучения особенностей образования гуанидинсодержащих сополимеров с виниловыми мономерами.
Учитывая сказанное, синтез и исследование свойств новых гуанидинсодержащих сополимеров открывает новые возможности для синтеза полимеров с необходимым набором свойств для конкретной области применения. В связи с этим цель данной работы заключалась в исследовании возможности получения новых высокомолекулярных сополимеров на основе АГ и МАГ с акриламидом (АА) и мономалеинатом гуанидина (ММГ) в водных растворах и, с учетом этих результатов, направленном синтезе новых полимеров катионной природы с биоцидными свойствами, изучении механизма и кинетики особенностей этих реакций. Для достижения поставленной цели необходимо было решить следующие задачи:
1. Исследование возможности получения новых сополимеров на основе АГ и МАГ с АА и ММГ и синтеза на их основе новых катионных полиэлектролитов.
2. Установление основных кинетических закономерностей радикальной сополимеризации АГ и МАГ с АА и ММГ в водных растворах, определение констант сополимеризации и характеристической вязкости.
3. Изучение влияния строения и свойств полимеризующихся частиц на кинетику и механизм радикальной сополимеризации.
4. Исследование физико-химических, бактерицидных, токсикологических и флоккулирующих свойств синтезированных мономерных и полимерных продуктов.
ЛИТЕРАТУРНЫЙ ОБЗОР
Глава I. Кинетические особенности реакций радикальной полимеризации акриловой и мет-акриловой кислот в водных и органических растворителях
1.1 Водные растворы. Изменение рН растворов добавлением NaOH
К числу ранних работ, посвященных исследованию реакции полимеризации метакриловой кислоты (МАК) в водных растворах, относятся труды, выполненные в 50-е годы группой английских ученых [1, 2], в которых авторы выявили влияние рН реакционного раствора на общую скорость полимеризации МАК, где в качестве инициатора использовалась Н2 О2. В указанных работах установлено, что в кислых средах при рН > 2 скорость полимеризации МАК сильно уменьшается с увеличением рН (симбатно измененялись и молекулярные массы (ММ) образующихся полимеров), а при рН > 5,5 полимеризация МАК не наблюдалась вообще.
Поскольку известно, что в исследованном диапазоне рН происходит увеличение степени ионизации мономера, Качальский и Блауэр высказали предположение [1, 2], что мономер МАК лишь в неионизованной форме способен участвовать в реакции роста цепей, т.е. «истинная» константа общей скорости полимеризации kр связана с наблюдаемой константой роста k соотношением kp = k/(1–a), где a – степень ионизации мономера. В других исследованиях [3, 4] авторы, использовавшие в качестве инициатора персульфат калия, показали, что полимеризация МАК в водных растворах происходит до высоких значений рН (около 13) [3]. Возможно, что Качальский и Блауэр не смогли установить это в более ранних работах вследствие того, что выбранный ими инициатор Н2 О2 был неэффективен при более высоких (больше 6) значениях рН.
Выявив, что метакрилатанион легко сополимеризуется при pH = 7 с N,N-диэтиламино-2-этилметакрилатом и акрилонитрилом и основываясь на этих данных, Пиннер с сотрудниками пришли к выводу, что падение скорости полимеризации МАК с ростом pH в интервале pH = 2,5-6 можно объяснить сополимеризацией неионизованной МАК и метакрилатаниона, и связали наблюдаемое падение скорости полимеризации при увеличении pH с возрастанием содержания метакрилатаниона в исходной смеси [4].
В более поздних исследованиях Блауэр и сотр. [5, 6], измеряя кинетику полимеризации МАК, инициированной динитрилоазобисизомасляной кислотой (ДАК), показали, что при pH> 6 полимеризация МАК также имеет место, и реакция при этом имеет первый порядок по концентрации мономера (при pH = 6; 9.5; 11). На основании этого авторы полагали, что первый порядок по мономеру сохраняется во всем интервале значений pH. Порядок скорости реакции по инициатору при pH = 10,6-11,6 оказался близким к 0,5. В щелочной области pH скорость полимеризации вначале несколько увеличивается с ростом pH, а затем снижается. Таким же образом изменяются и молекулярные массы полимеров. В этих работах впервые было установлено также, что при добавлении низкомолекулярных электролитов (нейтральных солей) в интервале pH = 6,8 – 9,6, скорость полимеризации ощутимо возрастает (табл. 1).
Таблица 1
Зависимость константы общей скорости полимеризации метакриловой кислоты от ионной силы раствора (61,4 °С)
| рН | Концентрация, моль/л | k´105, сек-1 | |
| мономер | cоль (NaCl) | ||
| 6,8 | 0,2 | - | 1,19 |
| 7,0 | 0,2 | 0,4 | 1,78 |
| 9,5 | 0,1 | - | 1,51 |
| 9,6 | 0,1 | 0,4 | 2,00 |
Для объяснения некоторого возрастания скорости полимеризации метакриловой кислоты в щелочной области Блауэр предположил, что с ростом рН уменьшается константа скорости бимолекулярного обрыва цепей из-за электростатического отталкивания заряженных макрорадикалов. Если это предположение верно, то увеличение ионной силы реакционных растворов при добавлении низкомолекулярных солей, способствуя столкновениям одноименно заряженных частиц, должно было бы привести к увеличению константы скорости бимолекулярного обрыва, т.е. к уменьшению общей скорости полимеризации. В то же время на опыте наблюдается обратное явление (табл. 1).
Кинетика полимеризации другой слабой непредельной кислоты – акриловой (АК) – в водных растворах изучалась японскими исследователями [7, 8]. И для этого мономера, как видно на (рис. 1), скорость полимеризации также зависит от рН реакционного раствора.
Авторами показано также, что увеличение ионной силы реакционного раствора при рН = 7,2, в результате добавления в него NaCl, как и в случае МАК, приводит к росту скорости полимеризации АК и увеличению ММ образующихся полимеров.
Наиболее полное и систематическое исследование кинетических особенностей и механизма реакций радикальной полимеризации АК и МАК в различных средах, в том числе в водных растворах, проводилось В.А. Кабановым, Д.А.Топчиевым и сотр. [9-21]. Ими было установлено, что в водных растворах во всем исследованном интервале рН (2,5-13,0) при использовании инициатора ДАК, как при полимеризации АК, так и для МАК, (рН раствора создавалось добавлением щелочи NaOH) сохраняется половинный порядок скорости реакции по концентрации инициатора, что свидетельствует о бимолекулярном механизме обрыва цепей. Первый порядок по концентрации мономера наблюдается при полимеризации МАК при рН > 2,5. В случае же акриловой кислоты порядок по мономеру оказался равен 1,5. Аналогичные порядковые закономерности при полимеризации АК в водных растворах отмечались также и другими авторами [22, 23].
Детальное исследование кинетических особенностей и закономерностей радикальной полимеризации АК и МАК в водных растворах показало, что зависимость скоростей полимеризации указанных мономеров от рН реакционного раствора имеют вид, указанный на рис. 2.
Так, при полимеризации АК и МАК в водных средах, в интервале рН = = 2,5-7,0 наблюдается значительное падение скорости полимеризации соответствующих мономеров. Значения ММ образующихся полимеров изменяются симбатно. В соответствии со значением констант ионизации для АК рka = 4,2, а для МАК рka = 4,32 (эффективная константа ПАК рka = 6,4, ПМАК рka = 7,0), можно заключить, что в области pH < 6 с увеличением pH реакционного раствора при полимеризации АК и МАК рост цепей происходит на незаряженных макрорадикалах поликислоты. В области рН > 6 зависимость общей скорости полимеризации акрилат- и метакрилатанионов от рН, как было показано на рис. 2, носит экстремальный характер. С ростом рН в области рН = 7-11 и 7-12 соответственно, наблюдается возрастание общей скорости полимеризации и ММ образующихся полимеров. Важно, что скорость инициирования в интервале рН = 8-10 как в присутствии, так и в отсутствие низкомолекулярных неполимеризующихся солей, практически неизменна [15, 16].
Таким образом, наблюдаемое увеличение общей скорости полимеризации акрилат- и метакрилатанионов при возрастании рН в щелочной области или при увеличении ионной силы при фиксированном рН, как выяснилось, действительно вызвано возрастанием отношения констант скоростей элементарных реакций роста и обрыва цепей, т.е. величины kр /kо1/2 .
На основании рассмотрения литературных и экспериментальных данных В.А.Кабанов и Д.А.Топчиев [14, 19-20], выдвинули гипотезу о кинетической роли ионных пар при радикальной полимеризации ионизирующихся мономеров. Для случаев полимеризации акрилат- и метакрилатанионов в водных растворах эта гипотеза была сформулирована и обоснована следующим образом. В водных растворах при рН > 7 АК и МАК полностью
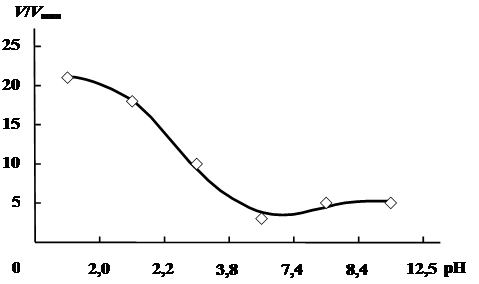 Рис. 1. Зависимость V/Vмин от рН при полимеризации АК в водных растворах.
Рис. 1. Зависимость V/Vмин от рН при полимеризации АК в водных растворах.
[АК] = 0,5 моль/л; [I]=2,85 10-3 моль/л; 50 °С

Рис. 2. Зависимость V/Vмин при полимеризации АК (1) и МАК (2) от рН
водного раствора, установленного добавлением NaOH. [АК] = 1,2 моль/л;
[ДАК] = 5×10-3 моль/л; Vмин = 4×10-6 моль/л×с; [МАК] = 0,92 моль/л;
[ДАК] = 5×10-4 моль/л; Vмин =1,15×10-6 моль/л×с; 60 °С.
ионизованы, т.е. в реакционном растворе присутствуют только акрилат- и метакрилатанионы. ПАК и ПМАК в среднем значительно слабее соответствующих мономерных кислот и их ионизация с ростом рН происходит в щелочной области, т.е. наблюдаемым в этой области рН кинетическим эффектам сопутствует изменение химической природы растущих цепей. Возможность изменения эффективной реакционной способности макрорадикалов как раз и предусматривается данной гипотезой. Предполагается, что если ионизованные, т.е. отрицательно заряженные, макрорадикалы роста способны образовывать ионные пары с низкомолекулярными катионами, присутствующими в растворе, в частности, на концах растущих цепей, то это должно приводить к возрастанию скорости реакции роста цепи из-за снятия электростатического отталкивания между растущим радикалом и одноименно заряженным мономером. Тогда должно нивелироваться электростатическое отталкивание при сближении мономерных анионов с одноименно заряженными радикалами в актах роста цепи [19, 20].
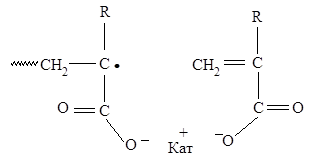
Схема 1
С ростом рН должна увеличиваться вероятность образования ионных пар на концах растущих цепей, что в рамках гипотезы должно приводить к увеличению эффективной константы скорости роста kр. Действительно, в соответствии с данными работ [15, 16], повышение концентрации ионов Na+ при данной концентрации мономера и фиксированном значении рН раствора (рН > 7) сопровождается увеличением общей скорости полимеризации [16]. (Понятие «ионная пара» используется в терминах Фуосса, т.е. предполагается непосредственный контакт сольватных оболочек противоположно заряженных ионов [20]).
Хорошо известно, что полианионы поликислот способны связывать низкомолекулярные противоионы в водных растворах [24]. Обычно сродство между противоположно заряженными низкомолекулярными ионами, например между Na+ и CH3 COO–, в не слишком концентрированных водных растворах недостаточно для стабилизации ионных пар. Образование стабильных ионных пар наблюдается лишь в менее полярных органических растворителях. Однако в случае полиэлектролитов связыванию способствует электростатическое поле всего полииона. Поэтому при высоких степенях ионизации значительная часть противоионов оказывается иммобилизованной в полимерных клубках.
Характерно, что в условиях, когда растущие цепи не ионизованы, добавление низкомолекулярных солей, как показали кинетические исследования, не влияет на скорость полимеризации акрилат- и метакрилатанионов (табл. 2) [15, 16].
Таблица 2
Кинетические эффекты, наблюдаемые при добавлении низко-молекулярных солей при радикальной полимеризации акрилат- и метакрилатанионов (60 °С)
| Акрилатанион [М]=1,2 моль/л, [ДАК] = 5´10–3 моль/л | Метакрилатанион [М] = 0,92 моль/л, [ДАК] = 4,85´10–4 моль/л | |||||
| рН* | Низкомолеку-лярная соль | V×105, моль/л×сек | рН* | Низкомоле-кулярная соль | V×105, моль/л×сек | [h]** |
| 8,0 | – | 1,9 | 9,5 | – | 0,20 | 0,62 |
| 8,0 | [NaCl]=2 моль/л | 7,5 | 9.5 | [КCl] = 0,9 моль/л | 1,61 | 1,4 |
* рН установлен добавлением NaOH; ** в 0,02 N водном растворе NaOH.
Для получения прямой информации о влиянии рН и ионной силы раствора на константы скоростей элементарных стадий процесса, в работе [15] было проведено детальное кинетическое исследование фотополимеризации метакрилат- и акрилатанионов в водных растворах, рН которых устанавливали добавлением NaOH. Результаты проведеных исследований и измерений представлены в табл. 3 и 4.
Таблица 3
Константы скоростей элементарных реакций при полимеризации крилатаниона
| рН | V´106 моль/л с | Vин´109 моль/л с | kp/k01/2, л1/2/моль1/2сек1/2 | k0´10–8, моль/л с | kр, моль/л с |
| 7,9 | 5,1 | 9,1 | 0,04 | 2,6 | 650 |
| 7,9* | 25,0 | 9,1 | 0,20 | 2,6 | 3100 |
| 11,0 | 51,0 | 9,1 | 0,40 | 2,7 | 6600 |
| 13,6 | 21,0 | 9,1 | 0,17 | 2,8 | 2500 |
[М] = 1,2 моль/л; [ДАК] = 8,3´10-4 моль/л; 23 °С.
*) 1,5 н р-р NaCl в воде
Таблица 4
Константы скоростей элементарных реакций при
полимеризации метакрилатаниона
| рН | V´106, моль/л с | Vин´109, моль/л с | kp/k01/2, л1/2/моль1/2сек1/2 | k0´10–8 л/моль с | kр, л/моль с |
| 8,0 | 4,2 | 8,9 | 0,046 | 2,1 | 670 |
| 13,6 | 12,0 | 8,9 | 0,130 | 2,3 | 1900 |
[М] = 0,92 моль/л; [ДАК] = 2,5´10-4 моль/л; 23 °С.
Как видно из данных, представленных в этих таблицах, с повышением рН от 8 до 13,6 константа скорости роста kр возрастает примерно во столько же раз, во сколько и стационарная скорость процесса. Константа скорости обрыва при этом практически не изменяется, т.е. в согласии с гипотезой возрастание скорости полимеризации акрилат- и метакрилатанионов в щелочной области рН действительно вызвано увеличением константы скорости роста kр .
Практическое постоянство констант скорости бимолекулярного обрыва с ростом рН и при увеличении ионной силы раствора в щелочной области, установленное при полимеризации акрилат- и метакрилатанионов, можно объяснить, по мнению авторов, только допустив, что обрыв лимитируется диффузионными процессами. Поскольку гидродинамические размеры и, следовательно, скорость поступательной диффузии самих клубков заметно меняются в изученных интервалах изменения свойств реакционной среды, можно полагать, что лимитирующей стадией является диффузия друг к другу концевых звеньев двух макромолекулярных клубков, предварительно сблизившихся, согласно теории Норта [25, 26], на расстояние, меньше критического.
В кислой области (рН < 6) при полимеризации АК и МАК рост цепей ведут незаряженные макрорадикалы поликислот и с ростом рН возрастает содержание ионизованного мономера – акрилат- и метакрилатаниона соответственно. Для выяснения причин наблюдаемого резкого падения общей скорости полимеризации МАК с ростом рН было оценено относительное изменение константы скорости роста цепи (использовали метод передачи цепи в присутствии меркаптоэтанола) и показано, что установленное падение общей скорости полимеризации обусловлено уменьшением константы скорости элементарной реакции роста цепи kр [21].
В результате измерений макроскопической вязкости модельных растворов, было установлено [21], что при изменении рН в интервале значений 2,5-5,5 гидродинамичекие размеры полимерных цепей, а, следовательно, и макрорадикалов не меняются заметным образом, следовательно не происходит изменения конформационного состояния растущих цепей в рассмотренном диапазоне рН. Вместе с тем, при повышении рН, в исследованных условиях, происходит ионизация МАК и меняется соотношение в исходном растворе двух, по существу различных мономеров: метакриловой кислоты и метакрилатаниона, способных присоединяться к неионизованным радикалам роста ПМАК. На основании данных, представленных в работе [27] (полученных при исследовании процесса сополимеризации МАК в водных растворах с другими сомономерами), известно, что МАК и метакрилатанион характеризуются различными значениями параметров реакционной способности Q и e. Поскольку концентрация метакрилатаниона в исследованном диапазоне рН увеличивается с ростом рН и считая (в соответствии с величиной константы ионизации МАК), что при рН 2,5 МАК полностью неионизована, а при рН 5,5 в реакционном растворе присутствует исключительно метакрилатанион, можно рассчитать отношение констант скоростей присоединения МАК и метакрилатаниона к неионизованному радикалу роста в рамках схемы Q-e :
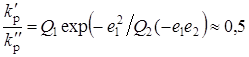 ,
,
где Q1 =2,34; e1 = 0,65 – константы для МАК.
Q2 = 1,36; e2 = –1,18 – для метакрилатаниона.
В действительности же, по данным работы [21], как видно, отношение констант равно лишь 0.5, а скорость полимеризации МАК в интервале рН = 2,5-5,5 падает примерно в 5 раз, т. е. экспериментальный результат не удается объяснить только различием классических параметров реакционной способности «сомономеров».
Причина наблюдаемого резкого уменьшения константы скорости роста цепей с увеличением рН от 2 до 6, как считают авторы [19-21], состоит в том, что по мере ионизации МАК концентрация сильно гидратированных метакрилат-анионов в относительно гидрофобных незаряженных клубках макрорадикалов ПМАК оказывается ниже их средней концентрации в растворе и, в то же время, в кислых средах можно ожидать преимущественной сорбции МАК в клубках ПМАК, т.е. повышения концентрации мономера в микрообластях, окружающих активные центры.
Интересные результаты о существенном влиянии природы катиона на эффективность константы сополимеризации в системе АК – акриламид в водных растворах приводились в работе Войнаровского и др. [28, 29], которые показали, что содержание звеньев акриловой кислоты в сополимере уменьшаются в ряду Li+ > Na+ > K+ .
Обнаруженные эффекты авторы связали с электростатическими взаимодействиями заряженных макрорадикалов и противоионов (катионов металлов), отметив при этом, что чем сильнее заряженные макрорадикалы (звенья акрилатанионов) способны к связыванию с катионами металла, тем в меньшей степени электростатические отталкивания препятствуют присоединению акрилатанионов в актах роста цепей.
Ранее Крещенцев и др. [20] показали, что при связывании «мертвой» полиакриловой кислоты с ионами металлов степень ионного связывания изменяется в таком же порядке Li+ > Na+ > K+, уменьшаясь с возрастанием радиуса катиона.
Регулирование рН растворов добавлением аминов. Особый интерес представляют данные по изучению влияния на процесс полимеризации рассматриваемых мономеров различных нейтрализующих агентов, в том числе органических аминов, основность и строение которых можно широко варьировать.
Основные результаты исследования таких систем были получены в работах Топчиева, Кабанова и сотр. [9-20]. В качестве нейтрализующих агентов были выбраны при проведении кинетических исследований полимеризации МАК – изобутиламин (ИБА), этилендиамин (ЭДА), триэтиламин (ТЭА) и гидроокись аммония; при полимеризации АК – ТЭА и NH4 OH. Было установлено, что во всех указанных системах сохраняется половинный порядок по инициатору (ДАК). По мономеру — при полимеризации МАК — установлен первый порядок, а полуторный порядок, при полимеризации АК. Было также показано, что скорость распада инициатора ДАК при полимеризации МАК не зависит от природы нейтрализующего агента и практически постоянна в исследованном интервале значений рН реакционных растворов [15].
Учитывая обнаруженную ранее особенность поведения акриловой кислоты, которая в отличие от МАК, характеризуется относительно высокой способностью к присоединению первичных и вторичных органических аминов по реакции Михаэля [30-32], при изучении полимеризации АК в воде в зависимости от рН был выбран ТЭА, влияние которого на кинетику полимеризации сравнивали с влиянием аммиака [15, 16, 19, 20].
Ход зависимости полимеризации АК и МАК от рН реакционного раствора имеет сложный характер. В области рН = 2-4,5 природа нейтрализующих агентов, как видно, практически не влияет на скорость полимеризации, и их роль, по мнению авторов [20], сводится только к ионизации мономера. С ростом рН происходит увеличение содержания метакрилат(акрилат)аниона (причины падения скорости полимеризации АК и МАК с ростом рН уже были обсуждены выше), а растущие полимерные цепи в рассматриваемом интервале рН не ионизованы.
При больших рН (5-10) зависимость скорости полимеризации МАК в случае добавления в реакционную систему ЭДА или NH4 OH проходит через экстремум. Для системы МАК–ЭДА максимум скорости наблюдается при рН = 6,6, когда макрорадикалы ПМАК преимущественно еще не ионизованы (константы диссоциации ЭДА равны: рКa1 = 7,0; рКа2 = 10,0). Авторами было высказано предположение, что неионизованные аминогруппы ЭДА, присутствующие в реакционных растворах, в этих условиях способны, по-видимому, образовывать ассоциаты с неионизованными карбоксильными группами на концах растущих цепей. В таком случае переходное состояние в акте роста цепи для данной системы можно представить следующим образом (схема 2).
Поэтому можно ожидать, что электростатическое притяжение протонированной (второй) аминогруппы ЭДА и метакрилатаниона должно дополнительно способствовать присоединению метакрилатаниона к активному центру, связанному водородной связью с неионизованной аминогруппой ЭДА. Существенный вклад в стабилизацию таких ассоциатов могут также вносить гидрофобные взаимодействия. Однако, при переходе к нейтральным рН (6,8-7,5) концентрация подобных ассоциатов, очевидно, уменьшается (вследствие ионизации самих карбоксильных групп в звеньях растущих цепей), что приводит к падению скорости полимеризации.
Схема 2
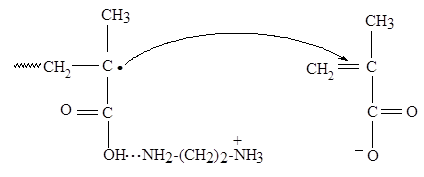
В щелочной области рН присутствие ЭДА и ИБА (рК = 10,4) приводит к более резкому, чем при добавлении NaOH, возрастанию скорости полимеризации метакрилатаниона с ростом рН. В рамках гипотезы о кинетической роли ионных пар это можно объяснить тем, что при рН > 7,5, когда макрорадикалы преимущественно ионизованы, происходит образование ионных пар на концах растущих цепей с участием карбоксилатанионов макрорадикалов и органических катионов, что способствует ускорению реакции роста цепи. В этих случаях, видимо, образованию ионных пар с участием этилендиаммония и изобутиламмония способствуют и гидрофобные взаимодействия, дополнительно стабилизирующие ионные пары.
По всей вероятности, гидрофобные эффекты ответственны также и за наблюдаемые значительные отличия в кинетике полимеризации акрилат- и метакрилатанионов в водных растворах, рН которых установлен добавлением триэтиламина. Существенно бóльший кинетический эффект с ростом рН при полимеризации метакрилатаниона авторы связали с установленными ранее доказательствами того, что для цепей ПМАК гидрофобные взаимодействия существенно выше, чем для ПАК [33] и предположили, что именно такие взаимодействия дополнительно стабилизируют ионные пары на концах ионизованных радикалов роста ПМАК.
Интересные результаты были получены в ходе кинетических исследований полимеризации рассматриваемых кислот при использовании в качестве нейтрализующего агента слабого неорганического основания NH4 OH. Авторами было установлено, что, как при полимеризации АК, так и для МАК наблюдается более резкое увеличение скорости полимеризации. Особенно значительно это увеличение проявляется при полимеризации метакрилатаниона.
Обнаруженные различия в скоростях полимеризации МАК и АК в области рН = 6-9 в присутствии NaOH и NH4 OH [10, 12] авторы объяснили специфичностью связывания ионов Na+ и NH4+ с ПМАК, что вытекает из рассмотрения кривых титрования соответствующих поликислот [19]. Существенно больший кинетический эффект (с ростом рН) при полимеризации метакрилатаниона также связали с ярко выраженными гидрофобными свойствами ПМАК (по сравнению с ПАК), предположив, что и в случае добавления NH4 OH в реакционные растворы так же, как и в случае ЭДА и ИБА, гидрофобные взаимодействия должны дополнительно стабилизировать ионные пары на концах ионизованных радикалов ПМАК.
В работах этих же авторов [10,12] был обнаружен также качественно другой тип влияния нейтрализующего агента при полимеризации МАК в водных средах, когда рН реакционного раствора создавался добавлением в него слабого органического основания – пиридина (Py) (рКа – 5,19). Полимеризация метакриловой кислоты от рН в присутствии пиридинийионов заметно отличается от аналогичной зависимости, обнаруженной в растворе NaOH и других изученных нейтрализующих агентов – ЭДА, ИБА, NH4 OH. Во всех указанных случаях наблюдали падение скорости полимеризации с ростом рН до 4,5.
Характерной особенностью обнаруженной зависимости является наличие острого максимума в интервале рН = 3,5-4,0, причем скорость реакции оказывается на порядок выше скорости полимеризации метакриловой кислоты в растворе NaOH при тех же значениях рН ([МАК] = 0,92 моль×л-1 ). Увеличение скорости происходит даже тогда, когда в растворах присутствует относительно небольшое количество пиридина. Для обеих концентраций максимальной скорости полимеризации соответствует исходное молярное отношение [Ру]/[МАК] = 0,23.
В качестве вероятных причин объясняющих аномальный ход кривой скорости полимеризации МАК, авторы рассматривают возможное влияние присутствующих в водном растворе линейных димеров МАК, о которых сообщалось в ряде работ [34, 35]. При этом было установлено, что в водных растворах карбоновых кислот имеются линейные димеры (схема 3), тогда как в неполярных растворителях присутствуют циклические димеры [36].
Схема 3
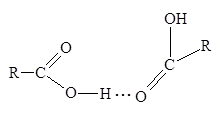
Увеличение скорости полимеризации МАК при рН = 3-4, сопровождаемое помутнением системы и выпадением полимера, обуславливается образованием донорно-акцепторного комплекса между ионизованным димером МАК (в котором система сопряженных связей играет роль донора электронов) и ионом пиридиния (акцептором электронов), который стабилизован гидрофобными взаимодействиями (схема 4).
Схема 4
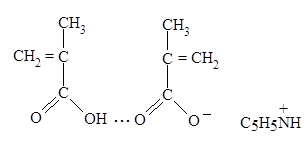
В образовавшемся комплексе происходит перераспределение электронной плотности на двойных связях, что может привести к химической активации димера. Увеличение рН раствора до 5-6 приводит к ионизации второй молекулы мономера входящей в димер и, следовательно, приводит к разрушению комплекса и к резкому падению скорости полимеризации. Константы ионизации таких димеров выше констант ионизации соответствующих мономерных кислот. Основная роль в стабилизации линейных димеров принадлежит гидрофобным взаимодействиям алкильных групп [35].
Роль гидрофобных взаимодействий в рассматриваемом процессе была показана также при изучении кинетики полимеризации другой непредельной кислоты АК в водных растворах, рН которых были установлены добавление NaOH и пиридина. Известно, что гидрофобность АК существенно менее выражена, чем гидрофобность МАК. Оказалось, что изменение скорости полимеризации АК в интервале рН = 2-6 мало зависит от природы нейтрализующего агента, т.е. в присутствии пиридинийионов не происходит ускорения полимеризации АК.
В результате проведенных исследований данного явления, авторы заключили, что в рассмотренной системе реализуется случай взаимодействия саморегулирующегося активатора – пиридинийиона – с димером МАК, в результате чего образуется комплекс с более высокой реакционной способностью, чем у МАК.
Таким образом рассмотренные в данном разделе результаты изучения кинетики полимеризации метакрилат- и акрилатанионов в водных растворах оснований и органических аминов свидетельствуют о весьма слабом влиянии природы нейтрализующего агента на зависимость начальной скорости полимеризации непредельных кислот от рН реакционного раствора. В соответствии с выдвинутой гипотезой авторы [20] рассматривают это явление как результат специфичного влияния природы ионных пар на константу скорости роста цепи при полимеризации и растущий радикал ионизованной поликислоты-поликатиона, либо образованием комплекса ионизованных мономеров в водных средах (например, система МАК – пиридин).
1.2 Полимеризация непредельных кислот в водных и органических средах
В предыдущих разделах рассматривались основы кинетических закономерностей и особенностей реакции радикальной полимеризации мономеров акрилового ряда в водных растворах с изменением рН в присутствии различных нейтрализующих агентов. Анализ представленных литературных данных позволяет заключить, что обнаруженные кинетические особенности, главным образом, являются следствием специфических взаимодействий заряженных макрорадикалов и присутствующих в реакционном растворе низкомолекулярных противоионов.
Представляется также несомненно важным оценить влияние природы реакционной среды на процесс полимеризации рассматриваемых мономеров, в частности провести сравнительный анализ кинетических данных при полимеризации акриловых кислот в органических растворителях и в водных растворах. Данной проблеме посвящено значительное количество публикаций [37-43].
Известно, что при радикальной полимеризации непредельных кислот в органических растворителях важную роль играет электронодонорная способность растворителей, а, следовательно, степень сольватации мономера в реакционной системе. Для димерной формы непредельных кислот характерно большее значение Q1, т.е. большая реакционноспособность в рамках схемы Q–е, поскольку димерная форма характеризуется большей энергией стабилизации двойной связи по сравнению с мономерной.
Наиболее детальные количественные исследования об активирующей роли воды и Н-ионов на процесс полимеризации непредельных кислот и амидов принадлежат Абкину и сотр. [37-43].
В представленных работах авторы исследовали полимеризацию АК, МАК и фторакриловой кислоты (ФАК), инициированную фотохимическим распадом ДАК в следующих растворителях: воде, диметилформамиде (ДМФА), диметилсульфоксиде (ДМСО) (табл. 5).
Таблица 5
Параметры, характеризующие реакции роста и обрыва цепей при полимеризации МАК и АК в различных растворителях
(УФ, l = 365 нм, 20 °0С)
| Раство-ритель | Мономер | kр × 10-3 , л/(моль×с) | Ер , ккал/моль | Ар ×10–7 , л/(моль×с) | k0 × 107 , л/(моль×с) | Е0, ккал/моль | А0×10-7 , л/(моль×с) |
| Н2 О | АК ФАК МАК | 27,2 26,0 4,1 | 3,1 4,5 4,3 | 0,6 0,6 0,67 | 18 8,7 1,1 | 0,6 0,2 | 18 20 1,6 |
| ДМФА | АК ФАК | 4,2 2,2 | 5,6 4,9 | 6,2 0,75 | 10 7,4 | 0,4 | 10 15 |
| ДМСО | АК ФАК МАК | 0,5 0,85 0,15 | 8,0 5,2 7,6 | 47 0,66 4,1 | 2,0 4,9 0,9 | 0,4 1,0 | 2,0 10,0 4,8 |
Как следует из табл. 5, при переходе от водных растворов к растворам ДМФА и, особенно ДМСО, и константа скорости роста цепи kр, и константа обрыва цепи k0заметно уменьшаются (одновременно падает и скорость полимеризации).
Установлено, что данное явление не связано с увеличением диэлектрической проницаемости реакционной системы [44], поскольку при одинаковой концентрации воды скорость полимеризации в различных средах близка, хотя диэлектрическая проницаемость различается в весьма широких пределах.
Наибольшее изменение константы роста kр наблюдается при переходе от воды к ДМСО для АК (в 50 раз) и наименьшее для ФАК (в 30 раз). Обнаруженное влияние природы растворителей на кинетику радикальной полимеризации рассматриваемых непредельных кислот, по мнению авторов этих работ, связано со способностью конкретного растворителя разрушать димеры мономерных кислот и тем самым изменять их реакционную способность. Однако это предположение не представляется достаточно обоснованным, поскольку, как известно, содержание димеризованных молекул АК в ДМСО не превышает 10% (согласно криоскопическим измерениям). В воде АК находится в основном в виде отдельных молекул (концентрация димерной формы незначительна), то же относится и к ФАК, которая не обнаруживает склонности к ассоциативным взаимодействиям.
Поэтому только образованием димеров между молекулами акриловых кислот нельзя объяснить наблюдаемые кинетические эффекты при полимеризации акриловых кислот в различных растворителях. Влияние природы растворителя может быть связано с рядом причин, среди которых в первую очередь следует отметить следующие:
1. Образование донорно-акцепторных комплексов между полимерными радикалами и молекулами растворителя, приводящих к уменьшению реакционной способности радикалов.
2. Сольватация молекул мономера и полимерных радикалов или специфическое взаимодействие этих частиц с молекулами растворителя, в частности, за счет образования водородных связей, приводящих к изменению плотности электронов на С=С-связи мономера или неспаренного электрона в радикале, а следовательно, к изменению реакционной способности реагирующих частиц.
3. Изменение конформационных характеристик полимерных молекул в растворителях, обладающих различной растворяющей способностью по отношению к полимеру.
Отличительной особенностью полимеризации водорастворимых мономеров является гидрофобное взаимодействие, возникающее в водных растворах полимеров. Оно приводит к резкому различию кинетических параметров полимеризации этого класса мономеров в воде и растворах органических растворителей. Гидрофобное взаимодействие также обуславливает значительное уменьшение скорости полимеризации и величины kр при добавлении к воде уже небольших количеств органического растворителя вследствие предпочтительной сорбции органического растворителя полимерным клубком. Естественно, что образование межцепных ассоциатов в результате гидрофобных взаимодействий, а, следовательно, и их «каталитическое» воздействие должно зависеть от гидрофобности используемых мономеров.
Авторы рассматриваемых работ считают, что определенный вклад в наблюдаемые кинетические эффекты вносят и факторы, связанные с конформационным состоянием растущих цепей в указанном ряду растворителей (т.е. влияние изменения конформационного состояния макрорадикала при изменении природы растворителя на кинетику процесса вследствие изменения доступности полимеризующихся частиц к активным центрам). Не отрицая, в принципе, возможности вклада этого фактора в кинетику полимеризации в рассматриваемых системах, считается целесообразным также обратить внимание на следующее. Как было отмечено выше, для водных растворов (табл. 5) характерны значительно более высокие константы роста и обрыва цепей (одновременно и более высокие скорости полимеризации). Эти же эффекты наблюдали и для некоторых других водорастворимых мономеров. Поэтому можно предположить, что причиной этого может быть большая сольватация (по сравнению с органическими растворителями), т.е. гидратация полимеризующихся частиц в водных средах. Разные мономеры могут характеризоваться разной степенью сольватации. Нетрудно допустить, что в рассматриваемом ряду мономеров именно для АК должна наблюдаться склонность полимеризующихся частиц – молекул мономера и макрорадикалов – к гидратации в водных растворах. Неудивительно, что при переходе от ДМСО к воде наибольшие кинетические эффекты обнаруживаются при полимеризации АК. При обсуждении вопроса о сравнительной реакционной способности рассматриваемых мономеров в различных растворителях, полезно было бы иметь соответствующие данные по сополимеризационной активности этих мономеров.
Интересные результаты при исследовании водородной связи в условиях гетерофазной полимеризации АК и МАК были получены Шапиро и сотр., когда полимеризация инициировалась радиационно-химически [45-47]. Оказалось, что растворители, способные к образованию водородной связи – диоксан, СН3 СООН, метанол, вода – мало влияют на скорость полимеризации АК в гетерофазной области, тогда как углеводородные растворители – толуол, гексан, являющиеся осадителями по отношению к ПАК, приводят к резкому падению скорости полимеризации АК и уменьшению длины образующихся цепей. При полимеризации МАК отмеченные выше эффекты проявляются в существенно меньшей степени.
Авторы предполагают, что растворители первой группы увеличивают время жизни линейных ассоциатов АК, сшивая их, а растворители второй группы (углеводородные) смещают равновесие в сторону димерной формы [45-47]. Вопреки мнению авторов, трудно предположить, что растворители, способные к образованию Н-связи и участию в смешанных ассоциатах, не разрушают, а упрочняют линейные ассоциаты АК и что уменьшение относительного количества линейных ассоциатов АК достаточно для снижения скорости полимеризации АК почти на порядок. Поэтому более вероятным представляется объяснить обнаруженные кинетические эффекты в терминах изменения констант бимолекулярного и мономолекулярного обрыва растущих цепей.
Приведенные выше результаты убедительно показывают, что природа растворителя – важный фактор, определяющий ход радикальной полимеризации водорастворимых мономеров. Влияние растворителя в случае водорастворимых мономеров существенно выше, чем в случае радикальной полимеризации таких малополярных мономеров, как, например, стирол. Особенно большое изменение как общей скорости реакции, так и величины kр наблюдается при переходе от полимеризации в органических растворителях к полимеризации в водном растворе.
1.3 Сополимеризация
Свободнорадикальной сополимеризацией АА, МАА и соответствующих N-замещенных амидов с другими мономерами получают линейные разветвленные и сшитые сополимеры, растворимые в воде или органических растворителях. Карбоцепные полиамидные гомо- и сополимеры превосходят соответствующие сложноэфирные аналоги по прочностным свойствам, имеют более высокие температуры стеклования, труднее гидролизуются. Показано также [48], что исходные амидные мономеры СН2 =CRCONR'R" отличаются от близких по строению сложных эфиров большей скоростью полимеризации.
Технология получения акриламидных сополимеров в основном такая же, как и гомополимеров. Однако сополимеризация АА или МАА с различными мономерами протекает, медленнее, чем гомополимеризация акриламидов, что может повлечь за собой повышение содержания в сополимерах остаточных мономеров, обычно являющихся токсичными. Нежелательным является также образование при сополимеризации полимеров с меньшей средней ММ, чем при гомополимеризации АА. Это обусловлено более высокими значениями константы передачи цепи kМ на сомономеры, чем на АА, для которого значение kМ очень незначительно.
Основные типы сополимеров
На основе акриламидов получен большой ассортимент как ионогенных (катионных и анионных), так и неионогенных сополимеров.
К наиболее распространенным водорастворимым катионным сополимерам относятся сополимеры АА с N-(диалкиламиноалкил)акрилатами и метакрилатами (в первую очередь, с NN-диметиламиноэтилметакрилатом) в нейтрализованной или квартернизованной форме. В последнее время стали привлекать внимание аналогичные сополимеры с N-(диалкиламиноалкил)-акриламидами. Сополимеры с N-(диме-тиламинопропил)метакриламидом превосходят сополимеры с диметиламиноалкилметакрилатами по устойчивости к гидролизу в щелочной среде.
Анионные сополимеры получают сополимеризацией АА или МАА, в первую очередь, с АК или МАК и их солями. Из МАА и МАК в промышленности получают сополимер «Метас», применяемый как защитный реагент в буровой технике и для других целей. Полимеры, макромолекулы которых состоят из элементарных звеньев амида и соли АК, или МАК, образуются и в результате гидролиза ПАА и ПМАА, I, а также при полимеризации АА и МАА в присутствии гидролизующих, агентов. Однако эти полимеры отличаются от сополимеров АА, полученных радикальной сополимеризацией, характером распределения элементарных звеньев в макромолекулах. Анионные сополимеры, водные растворы которых обладают повышенной устойчивостью к разделению фаз под действием двухвалентных металлов, синтезированы сополимеризацией АА с мономерами, в которых кислотная группа непосредственно не связана с винильной группой, например с 3-акриламидо-3-метилбутаноатом натрия и 2-акриламидо-2-метилпропансульфонатом натрия. Сополимеры N-н-алкилакриламида (алкильная группа – C8, С10, С12 ) и З-акриламидо-З-метилбутаноата натрия образуют водные растворы, вязкость которых не уменьшается под действием электролитов [49].
При сополимеризации 2-акриламидо-2-метилпропансульфокислоты со стиролом [50] и с 9-винилфенантреном или 1-винилпиреном [51] в органических растворителях получены полимеры, в состав которых входят как гидрофильные, так и гидрофобные сегменты, причем первые (в виде солей) обладают высокой способностью солюбилизировать вторые в воде. Эти сополимеры служат средой для фотосенсибилизированных реакций переноса электрона. Широко известны сополимеры АА с n-стиролсульфокислотой и ее солями [52].
Среди ионогенных акриламидных сополимеров все больший интерес представляют полиамфолиты. Так, сополимеризацией в воде АА с метакрилатом натрия, 5-винил-1,2-диметилпиридинийметилсульфатом и NN-метилен-бис-акриламидом получены набухающие и коллапсирующие полиамфолитные сетки [53, 54]. Полиамфолиты синтезированы из смесей мономеров, содержащих соли («сомономеры»), катион и анион которых имеют винильные группы, участвующие в сополимеризации, например, 3-метакрил-амидопропилметиламмоний, 2-акриламидо-2-метилпропансульфонат [55, 56].
На основе акриламидов получают различные неионогенные сополимеры. К ним относятся сополимеры АА или МАА с N-замещенными акриламидами, не содержащими или содержащими в заместителе функциональные группы, сополимеры, для получения которых используются только замещенные амиды, сополимеры АА и МАА с α, β-ненасыщенными нитрилами, сложными эфирами и другими мономерами.
АА сополимеризуют c N-n-алкилакриламидами (алкильная группа – C8, С10, С12 ) для получения «гидрофобно ассоциирующих» полимеров. Наличие в сополимерах всего 0,25 — 0,5% (масс.) звеньев вторых мономеров способствует сохранению или даже увеличению вязкости водных растворов полимеров при добавлении к ним электролитов [57, 58].
На основе АА и N-(1,1-диметил-3-оксобутил)акриламида получают сополимеры, предельные числа вязкости которых при нулевом сдвиге возрастают в результате добавления одно- и двухвалентных солей. Предположено, что этот эффект связан с наличием в макромолекулах циклов за счет образования водородных связей [59].
Для межмолекулярного сшивания полимеров на основе АА, замещенных акриламидов и других мономеров широкое применение находят N,N'-метилен-бис-акриламид, N,N' -метилен-бис-метакриламид и другие мономеры на основе АА, содержащие две и более полимеризующихся группы. С увеличением доли сшивающих агентов в смеси мономеров снижается степень превращения, при которой эти агенты вызывают образование геля.
На основе АА и акрилата натрия с применением аллилового эфира карбоксиметилцеллюлозы в качестве полифункционального сшивающего агента синтезированы гидрогели с большой степенью набухания (влагоабсорбенты), причем набухшие гидрогели имели хорошие деформационно-прочностные характеристики [60].
Для получения термореактивных ариловых и других полимеров в макромолекулы путем сополимеризации часто вводятся элементарные звенья N-гидроксиметакриламида или N-гидроксиметилметакриламида [61, 62]. Структурированию полимеров, содержащих N-гидроксиметиламидные группировки, способствует наличие в макромолекулах незамещенных амидных групп. При сополимеризации акрилонитрила и 0,5 — 0,7% N-гидроксиметил-метакриламида в отсутствие [63] или в присутствии 1-8% АА [64] образуются термически сшиваемые волокнообразующие сополимеры. При сополимеризации метилметакрилата, N-гидроксиметилметакриламида и N,N'-метилен-бис-метакриламида может быть получено модифицированное органическое стекло [61].
К новым направлениям синтеза сополимеров АА относится сополимеризация АА с макромономерами (Мn = 1100-4600) строения
СН2 =СНСООСН2 СН2 S(СН2 СН)n Н
|
СООС12 Н25
синтезированными теломеризацией додецилакрилата в присутствии 2-мер-каптоэтанола в качестве телогена с последующим ацилированием теломеров акрилоилхлоридом. При этом получены сополимеры с соотношением элементарных звеньев в основной цепочке 160: (2,5-1) [65].
Закономерности сополимеризации
Закономерности сополимеризации определяются, в первую очередь, строением исходных мономеров и средой, в которой проводится процесс. Оба фактора в полной мере проявляются при сополимеризации непредельных амидов. Для «классических» вариантов сополимеризации вклад этих факторов оценивается по их влиянию на скорость сополимеризации, степень полимеризации и относительные активности мономеров (константы сополимеризации) r1 и r2. При этом r1 =k11 /k12 и r2 =k22 /k21, где k11, k12 – константы скоростей реакций макрорадикала М1 с «собственным» (M1 ) и «чужим» (М2 ) мономерами; k22, k21 – константы скоростей реакций макрорадикала М2 с мономерами М2 и M1 .
Показателями активности мономеров при сополимеризации, как известно, являются также полуэмпирические параметры Q и е, предложенные Алфреем и Прайсом и характеризующие резонансный (наличие сопряжения) и полярный эффекты соответственно. Необходимо отметить, что многие реальные процессы полимеризации и сополимеризации с участием АА и замещенных акриламидов; являются осложненными («особыми») процессами [66, 67]. Поэтому приводимые значения r1, r2, Q1, Q2, е1, е2, k11, k12, k22, k21 часто представляют собой усредненные (эффективные) величины.
Влияние строения акриламидов на их реакционную способность при сополимеризации. Реакционная способность замещенных АА изменяется в широких пределах в зависимости от природы заместителей. Влияние последних выражается в виде полярного, резонансного и стерических эффектов. Рассматривая сополимеризацию в ряду замещенных непредельных амидов, удается вывести закономерности влияния отдельных эффектов и в тех случаях, когда одновременно значительное влияние оказывают и другие эффекты.
При исследовании радикальной сополимеризации АА с МАА было найдено, что при 25 °С r1 = 0,74 ± 0,11 и r2 = 1,1 ± 0,2. Несколько большую реакционную способность второго мономера связывают с тем, что замещение а-водородного атома в АА метальной группой приводит к повышению стабильности переходного состояния за счет сверхсопряжения. Вместе с тем, при взаимодействии с одним и тем же мономером метакриламидный радикал значительно менее реакционноспособен, чем акриламидный.
В данном случае определяющую роль, играет стерический эффект [68], При взаимодействии с ММА-радикалом N-арилметакриламид также оказался более активным, чем имеющий тот же заместитель АА [69].
При сополимеризации замещенных акриламидов CH2 =CHCONR'R" с АН в среде ДМФА величина r1 уменьшается в том же ряду, в котором изменяется скорость гомополимеризации этих же амидов (даны R' и R")[70]:
Н, СН3 > Н, Н > Н, н-С4 Н9 > С6 Н5, С6 Н5 ≥ СН3, СН3 .
Реакционные способности пара-замешенных N-фенилметакриламидов (1/r2 ) при сополимеризации в массе этих мономеров с ММА (М2 ) также убывают с уменьшением электронодонорной и увеличением электроноакцепторной способности пара-заместителя [71]:
СН3 О > СН3 > Н > Сl.
При изучении сополимеризации N-замещенных метакриламидов с АН были установлены линейные зависимости реакционных способностей lg(l/r2 ) 4-замещенных N-фенилметакриламидов от о-констант Гаммета и N-алкил-метакриламидов и N-фенилметакриламида от о-констант Тафта. Константы, характеризующие резонансный (BR) и стерический (Es) эффекты в уравнениях Гаммета и Тафта, не оказывали заметного влияния на значение 1/r2, т.е. изменение реакционных способностей рассматриваемых мономеров зависит в основном от полярного эффекта заместителей. Малые абсолютные значения р (–0,13) и р* (–0,033) в уравнениях Гаммета и Тафта характерны для гемолитических реакций. Отрицательные же значения этих констант, как и константы р* для реакции N-монозамещенных амидов с метилметакри-латным радикалом [72], связаны с тем, что при переходе к амиду с более электроноакцепторным заместителем уменьшается его реакционная способность по отношению к акрилонитрильному или метилметакрилатному радикалам, у которых заместитель также является электроноакцептором [70]. Следует отметить, что в ИК-спектрах N-моноэамещенных амидов полосы поглощения nС=С и nС=О смещаются в сторону более длинных волн с ростом электронодонорных свойств заместителей [72].
При изучении бинарной сополимеризации 1-акриламидо-1-дезокси-D-глюцита и 1-дезокси-1-метакриламидо-D-глюцита с различными виниловыми мономерами найдено, что при использовании винилацетата в качестве сомономера решающую роль играет наличие резонансной стабилизации в молекуле первого мономера и ее отсутствие во втором (r1 > r2 ); в случае же, когда оба мономера являются сопряженными (М2 – СТ, ММА), способность к сополимеризации определяется главным образом тем, что в первом мономере стерические препятствия играют значительно большую роль, чем во втором (r1 << r2 ) [73].
Константы сополимеризации N-акрилоилпирролидрна со СТ в бензоле (60 °С) оказались равными 1,5 и 0,35. Вычисленные на основании этих данных значения Q = 0,42 и е =1,60 для N-акрилоилпирролидона указывают, что этот мономер является высокополярным, но не проявляет существенной тенденции к резонансной стабилизации (эффект сопряжения мал). Замена в указанной паре мономеров акрилоильного производного метакрилоильным производным того же лактама изменяет относительные активности мономеров (r1 < 1; r2 > 1), что, связано с возникновением в системе заметных стерических препятствий [74]. При сополимеризации N-метакрилоил-б-капролактама со СТ [75] эти препятствия еще более существенны, и поэтому r1 становится равной нулю (замещенный амид не подвергается гомополимеризации). Значение r2 = 1 в данной паре мономеров указывает, что отношение констант скоростей стирольного радикала с обоими мономерами в значительной степени определяется противоположной полярностью этих мономеров.
При исследовании сополимеризации N-(н-октил)акриламида, N-(1,1,3,3-тетраметилбутил)акриламида [76] и N-(н-октадецил)акриламида [77] с ММА и СТ найдено, что в этих системах r1 < 1 и r2 > 1, т.е. указанные замещенные акриламиды по реакционной способности уступают сомономерам. Близость r1 и r2 в указанных парах мономеров и парах N-(н-октадецил)акриламид – ММА (СТ) и н-октадецилакрилат – ММА (СТ) позволяет считать, что стерический эффект (препятствия, создаваемые алкильными группами) определяет реакционную способность акриламидов, имеющих длинные объемные заместители у азота.
Наличие двух заместителей у азота АА не препятствует ни гомо-, ни сополимеризации мономеров, но вызываемые этими заместителями стерические препятствия сильно сказываются на кинетических параметрах образования полимеров. Так, константы сополимеризации в ДМФА (60 °С) N,N-диметил- и N,N-дибутилакриламидов со СТ [78] равны соответственно 0,23 и 1,23; 0,32 и 1,65. В этих системах сопряженных мономеров вопреки противоположной полярности соединений стирольный радикал предпочтительно реагирует со СТ (r2 > 1), очевидно, вследствие пространственных затруднений в N, N-дизамещенных акриламидах. На основании констант сополимеризации ряда N,N-дизамещенных акриламидов и констант скоростей роста при гомополимеризации соответствующих мономеров были вычислены константы скорости взаимодействия замещенного амидного радикала с «чужими» мономерами (k12 ) и «чужих» радикалов с амидами k21 [79]. Оказалось, что k12 очень сильно зависит от природы заместителей в амиде. Например, при сополимеризации в массе (30 °С) с ММА для N-акрилоилзамещенных диметиламина, пирролидона и пиперидина значения k12 относятся как 66:14:1. Поскольку же значения k21 для всех трех N, N-дизамещенных амидов при взаимодействии с одним и тем же мономером имеют один порядок, можно сделать вывод, что убывание k12 обусловлено возрастанием стерических препятствий в амидном радикале, создаваемых заместителями у азота.
N,N-Диалкил- и N-алкил-N-арилметакриламиды, которые не подвергаются радикальной гомополимеризации [72, 80, 81], сополимеризуются с некоторыми сопряженными мономерами, например со СТ, ММА [278], АН [268], N,N¢ -метилен-бис-акриламидом [80, 82]. Однако получаемые при низких конверсиях сополимеры обеднены звеньями амидов по сравнению с их содержанием в мономерных смесях. Так, при сополимеризации N,N-диме-тилметакриламида и ММА в диоксане (80 °С) r1 = 0,175, r2 = 8,92 [83]. Преобладающий вклад стерического фактора в реакционную способность N, N-дизамещенных метакриламидов подтверждается тем, что N-метакрилоил-азиридин, в котором ограничена подвижность заместителей у азота (поскольку они входят в состав напряженного трехчленного гетероцикла), в отличие от указанных N,N-дизамещенных метакриламидов, подвергается не только со-, но и гомополимеризации по радикальному механизму [84]. Получены также сополимеры двух дизамещенных метакриламидов – N-метакрилоилпи-перидина и N-метакрилоиланабазина [85], N-заместители каждого из которых входят в состав гетероциклов.
Предположение о том, что устойчивость к гомополимеризации N,N-ди-замещенных метакриламидов обусловлена превышением температуры опытов над критической температурой полимеризации, опровергается тем, что N,N-диметилметакриламид не превратился в полимер под действием УФ-излучения и при –78 °С [70].
Сополимеризация с неионогенными мономерами. На закономерности сополимеризации большое влияние оказывают условия проведения процесса. Известно [86], что появление при сополимеризации границы раздела фаз даже при отсутствии межфазного взаимодействия часто приводит к изменению состава сополимера и отклонению процесса в целом от схемы Майо — Льюиса. При гомофазной сополимеризации, если мономеры не подвергаются диссоциации, ассоциации или специфической сольватации молекулами растворителя, и при соблюдении pяда других условий [87] процесс образования сополимеров описывается уравнениями, вытекающими из классической теории сополимеризации. Ниже рассматривается, в какой мере от схемы Майо — Льюиса отклоняется сополимеризация α, β-ненасыщенных амидов с неионогенными мономерами, а именно с мономерами, которые в условиях сополимеризации, как правило, не диссоциируют и проявляют слабую склонность к автоассоциации и взаимодействию с растворителем. В таких системах отклонения от указанной схемы определяются в основном строением акриламидного компонента.
Наиболее показательно для отклонения закономерностей сополимеризации от схемы Майо — Льюиса наличие зависимости r1 и r2 от природы растворителя. В ряде работ приведены данные по зависимости r1 и r2 от природы растворителя при сополимеризации АА и СТ [88-91]. Как видно из табл. 6, значения r1 уменьшаются, а r2 увеличиваются при переходе от бензола и 1,2-дихлорбензола к бензонитрилу, простым эфирам, ДМСО и спиртам.
Таблица 6
Относительные активности АА и СТ при сополимеризации в различных растворителях при 30 0С (10%-е растворы) [286-288].
| Среда | r1 | r2 | ∆ поглощения, см-1 | |||
| С=О | NHI | NHII | (NHI +NHII)/2 | |||
| Бензол * | 12,5 | 0,25 | 1 | 15 | 10 | 12,5 |
| 1,2-дихлорбензол | 12,5 | 0,25 | 5 | 5 | 5 | 12,5 |
| Безонитрил | 2,4 | 1,35 | 12 | 70 | 55 | 62,5 |
| Диоксан | 1,38 | 1,27 | 10 | 205 | 170 | 187,5 |
| Диметиловый эфир диэтиленгликоль | 1,09 | 1,40 | 10 | 190 | 206 | 198 |
| Этанол | 0,30 | 1,44 | 13 | 205 | 230 | 217,5 |
| 2-(2-метоксиэтокси)этанол | 0,27 | 1,66 | 22 | 205 | 230 | 217,5 |
| Вода-трет-бутанол | 0,78 | 2,93 | 28 | 205 | 230 | 217,5 |
| ДМСО | 0,16 | 2,01 | 17 | 245 | 235 | 40 |
| Метанол | 0,20 | 1,05 | 13 | 205 | 230 | 217,5 |
*В 1%-м растворе [289] r1 =9,14 ± 0,27; r2 =0,67 ± 0,08.
Приблизительно в той же последовательности увеличивается смещение полос NH амидной группы в ИК-спектре растворов АА в перечисленных растворителях в сторону более длинных волн по сравнению с поглощением в тетрахлориде углерода, отнесенным к бесконечно разбавленному раствору. Одновременно наблюдается и некоторое смещение полосы С=0, но по своему абсолютному значению оно значительно уступает смещению полос NH. Из этих данных вытекает, что зависимость r1 и r2 от природы растворителя связана в основном с образованием водородных связей между амидными атомами водорода и молекулами растворителей, а также диполь-дипольным взаимодействием между этими же соединениями. В отличие от указанных факторов, диэлектрическая постоянная и дипольный момент [88] не оказывают решающего влияния на изменение состава образующихся сополимеров. Отдаление атомов водорода от амидного атома азота приводит к увеличению его отрицательности, которая распространяется на всю молекулу амида и обусловливает смешение π-электронов группы СН2 = СН к метилену и удлинение углерод-кислородной связи [82]. Поскольку направления поляризации молекул AA и СТ противоположны, то уменьшение электроноакцепторной способности амидмой группы в АА должно приводить к некоторому сближению полярностей обоих мономеров и к уменьшению значений констант k12 и k21. Что касается k21, то при малой зависимости реакционной способности СТ от среды (k22 = const) ее уменьшение должно приводить к росту r2, что и имеет место. Судя по тому, что с увеличением, связывания молекул АА растворителем r1 уменьшается, можно предположить, понижение k12 сопровождается еще большим понижение k11 в частности, из-за роста стерических затруднений при столкновении специфически сольватированных мономера и радикала акриламида.
При сополимеризации АА и ММА в ДМСО и хлороформе добавка небольших количеств воды приводит к заметному увеличению r1 и мало влияет на r2, что связано с ускорением гомополимеризации АА (рост k11 ) и, вероятно, обусловлено сольватированием растущих цепей молекулами воды [92]. С другой стороны, при сополимеризации АА и N-винилпирролидона в воде частичная замена последней глицерином, способным к специфической сольватации АА, также приводит к значительному росту r1 и некоторому понижению r2. Так, с увеличением содержания глицерина в растворителе от 0 до 80 % (масс.) при 60 °С r1 увеличивается от 0,60 до 1,06; r2 падает от 0,17 до 0,11 [93]. Приведенные данные указывают на очень сильную зависимость r1 и r2 от природы растворители и сложный характер этой зависимости: одни и те же вещества в зависимости от природы системы в целом могут вызывать противоположные эффекты.
При изучении эмульсионной сополимеризации АА и этилакрилата найдено [94], что состав сополимера отличается в сравнимых условиях в растворе, а под действием добавок ацетона, этанола, диакианы и других растворителей он изменяется.
При сополимеризации МАА [95] и N-метилакриламида [96] со СТ и ММА наблюдается заметное влияние среды на значения r1 и r2, по характеру такое же, как при сополимеризаиии АА со СТ.
Таблица 7
Относительные активности N-(1,1-диметил-3-оксобутил) акриламида и СТ при сополимеризации в разных растворителях при 70 °С (с общей концентрацией мономеров-0,8 моль/л) [95]
| Среда | r1 | r2 |
| Бензол | 0,49 | 1,77 |
| Диоксан | 0,47 | 1,85 |
| Этанол | 0,30 | 1,80 |
Изучение сополимеризации N-(1,1-диметил-3-оксобутил)акриламида со СТ и ММА в разных растворителях показало (табл.7), что относительная активность второго мономера практически не зависит от реакционной среды, первого же в бензоле и диоксане несколько больше, чем в этаноле, т.е. наблюдается та же законономерность, что при сополимеризации АА со СТ, но она слабее выражена. Это может быть обусловлено как сравнительно большим объемом заместителя атома азота, так и тем, что в молекуле N-(1,1-диметил-3- оксобутил)акриламида и соответствующего радикала имеется внутримолекулярная Н-связь [97], вследствие чего схема 5 :
![]() Н---О
Н---О
![]() СН2 =СНСОN С-СН3
СН2 =СНСОN С-СН3
(СН3)2 С – СН2
и сольватация в спиртовой среде молекулами pacтворителями подавлена. Напомним, что такая сольватации приводит к резкому изменению k11 и r1 при сополимеризации незамещенного азота у АА.
Влияние природы растворителя на скорость изучено на примере системы АА — АН. В растворителях, способных образовывать автоассоциаты посредством водородных связей (вода; уксусная кислота, метанол, ДМФА), скорость полимеризации резко понижается при добавлении небольших количеств АН к АА. В растворителях, не способных к автоассоциации, но способных к сольватации (диоксан, ацетон, ацетонитрил), скорость образования сополимера постепенно уменьшается пропорционально доле АН в мономерной смеси, В инертных растворителях (н-гексан, бензол, толуол) скорость практически не меняется до достижения содержания АА в смеси мономеров в 40% (маcс.), а при дальнейшем обеднении смеси амидом процесс замедляется [10].
Для дизамещенных у азота акриламидов и метакриламидов, в амидной группе которых нет подвижных атомов водорода, активно участвующих в образовании различного рода ассоциатов и комплексов с молекулами среды, нехарактерна заметная зависимость реакционной способности от природы растворителя. N,N-дизамещенные амиды образуют сополимеры одинакового состава и одинакового распределения по составу при сополимеризации в массе и в различных растворителях [96, 98, 99]. Исключение могут составить протонные растворители. Природа растворителя не оказывает влияния на значения r1 и r2 при сополимеризации также N-монозамещенных акрил- и метакриламидов, если заместитель стерически препятствует незамещенному амидному атому водорода участвовать в образовании комплексов с молекулами растворителя. Например, значения r1 и r2 не зависят от природы растворителя при сополимеризации N-(н-октадецил) акриламида с ММА и СТ [77].
Зависимость констант сополимеризации незамещенных и многих монозамещенных у азота амидов от природы растворителей позволяет отнести системы, содержащие эти мономеры, к категории осложненных («особых») систем [86], не подчиняющихся классической теории сополимеризации Майо-Льюиса. Для таких систем схема Алфрея-Прайса неприменима, поскольку значения Q и е становятся неоднозначными [100, 86]. Например, для МАА в литературе приведены следующие значения Q и e: 1,46 и 1,24 [101], 0,88 и 0,74 [68], 0,57 и – 0,06 [71]. Очевидно, что пользоваться значениями Q и е как константами, характеризующими данный мономер, в случае соединений, обладающих значительной склонностью к ассоциации и к сольватации (особенно, специфической), не следует. При рассмотрении «особых» систем параметры Q и е могут служить лишь в качестве условных величин, отражающих влияние тех или иных факторов на поведение данного мономера при сополимеризации.
Более или менее стабильные значения Q и e могут быть характерны для N, N-дизамещенных амидов, а также для N-моноэамешенных амидов, у которых вследствие большого объема заместителей подавляется или резко ограничивается ассоциация мономера и доступ к амидным атомам водорода молекул растворителя. Постоянство Q и е в разных средах наблюдается при сополимеризации N,N-диметилакриамида с различными мономерами [102, 94-103], масляного эфира N-гидроксиметилметакриламида с ММА и АН, N-(н-октадецил)акриламидов с ММА и СТ [77]. Однако, учитывая значительный вклад стерических эффектов в реакционную способность N,N-дизамещенных амидов, а также то, что Q, е-схема не применима к системам, содержащим стерически сильно затрудненные мономеры, параметры Q и e рассматриваемых амидов не являются константами, характеризующими их резонансную стабилизацию и полярность.
Заслуживает специального внимания вопрос о зависимости значений r1 и r2 от конверсии мономеров. Было вполне естественно ожидать, что при сополимеризации мономеров, образующих «особые» системы, по мере увеличения содержания полимера в реакционной среде будет изменяться характер взаимодействия между компонентами смеси и, следовательно, будут изменяться значения относительных активностей мономеров. Данные по гомофазной и гетерофазной сополимеризации АА и АН [5 — 20 и 30 — 80% (мол.) соответственно] в водных растворах [104] полностью подтвердили эти ожидания. Для ряда степеней превращения были определены текущие отношения концентраций амида и нитрила в мономерной смеси (М1 /М2 = F) и соответствующие им отношения количеств мономеров (m1 /m2 = f), пошедших в состав сополимера в данный момент времени («мгновенный» состав сополимеров). Далее, пользуясь уравнением состава сополимера в форме, предложенной в работе [105], найденные зависимости изобразили графически. При всех соотношениях мономеров, независимо от того, выделялся сополимер в виде твердой фазы или нет, не были получены линейные зависимости (рис. 3).
Одновременно было показано [104], что найденные по начальным скоростям при 20 °С константы сополимеризации в гомофазной среде в отсутствие и в присутствии сополимера резко отличаются:
Без добавок сополимера 0,65 + 0,04 2,34 ±0,35
С добавкой сополимера 0,027± 0,003 1,45 ± 0,41
Рис. 3. Зависимость состава сополимера АА и АН от состава мономерной смеси в координатах уравнения Файнмена-Росса [300]при сополимеризации до глубоких степеней превращения (вода, 20 °С, начальные концентрации: АА – 0,42, АН – 0,95 моль/л)
Необходимо отметить, что основную ответственность за осложненный («особый») характер системы АА – АН несет первый мономер. На этo указывают результаты исследования гомофазной сополимеризации АН и СТ до глубоких степеней превращения, согласно которым относительные активности в ходе процесса меняются (r1 уменьшается) лишь при преобладании нитрила в смеси мономеров [83]. Кроме того, при сополимеризации ММА с N,N-диметилметакриламидом, в амидной группе которого нет атомов водорода, участвующих в образовании амидных ассоциатов переменного состава, значения r1 и r2 в ходе процесса оставались постоянными [106].
В ходе сополимеризации АА и МАА с ММА в растворах ДМСО относительная активность амидов падает, а эфира – растет[107]. Предположено, что для систем (мет)акриламида и мономера, не участвующего или слабо участвующего в образовании автоассоциатов или комплексов, изменение относительной активности мономеров обусловлено тем, что по мере протекания гомофазной сополимеризации уменьшается доля более активного амида, входящего в состав автоассоциатов этого мономера, и увеличивается доля менее активного мономера, образующего смешанные ассоциаты с акриламидными звеньями сополимера.
На примере системы МАА — ММА была предложена методика количественной оценки изменения относительных активностей мономеров при сополимеризации: использование способа Келена и Тюдоша [108] для определения r1 и r2 по данным среднего состава сополимеров при глубоких степенях превращения позволило определить изменяющиеся «интегральные» значения r1 и r2, достигаемые при каждой степени превращения мономеров в сополимер (при близких конверсиях в различных сериях опытов). Для рассматриваемой системы найдено, что при конверсии до 32% r1 постепенно уменьшается от 0,50 до 0,26, а r2 увеличивается от 4,2 до 5,0. При оценке относительных реакционных способностей в системе АА – СТ на основании данных о составе сополимера при высоких степенях превращения [109] в различных растворителях получены значения [110], заметно отличающиеся от найденных при малых конверсиях [91]. Значения, найденные в работе [110], можно отнести к интегральным r1 и r2 .
Обратим внимание еще на одну особенность сополимеризации aмидсодержащих систем, которые можно отнести к «особым». В тройных системах [111], в состав, которых входят амиды, имеющие склонность к образованию различного рода ассоциатов, реакционная способность компонентов отличается от их реакционной способности в соответствующих бинарных системах, причем направление и степень отклонений зависят от характера межмолекулярных взаимодействий. Очевидно, природа ассоциатов, образованных в растворе двумя соединениями, может изменяться при появлении в системе третьего соединения. В связи с этим использование метода Алфрея и Голдфингера [112] вычисления составов тройных сополимеров на основании значений r1 и r2 соответствующих трех бинарных систем для амидсодержащих систем может давать результаты, заметно отличающиеся от экспериментальных. Это положение экспериментально подтверждено на примере тройных смесей мономеров, содержащих наряду с амидом также кислоту или аммониевую соль. Для системы АА – АН – МАК уже при малых степенях превращения характерно большее обогащение сополимеров нитрилом и кислотой, чем это следует израсчета (рис. 4).
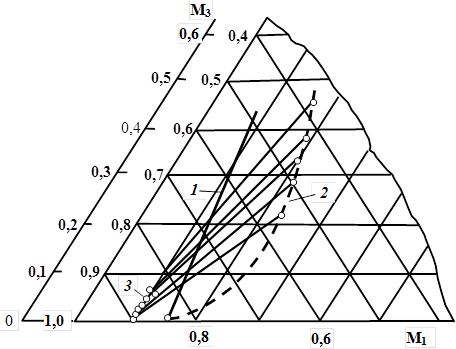
Рис. 4. Зависимость вычисленного (1) и найденного экспериментально (2) состава терполимера от состава мономерной смеси (3) в системе АА (М)1 — акрилонитрил (M2 ) — метакриловая кислота (M3 ) [307]
В системе МАА – гидрохлорид N,N-диэтиламиноэтилметакрилат-2-гидроксиэтилметакрилат получаемый сополимер содержал звеньев второго мономера меньше, а третьего – больше, чем по расчету [111].
При радикальной сополимеризации N-н-оксиакриламида и N,N-ди-бутилакриламида со СТ в среде толуола (25 °С) в присутствии этилалюми-нийсесквихлорида в качестве комплексообразователя получаются чередующиеся сополимеры [113].
Сополимеризация с ненасыщенными кислотами и их солями. Важной особенностью сополимеризации АА с мономерами, содержащими свободную или нейтрализованную кислотную группу, например, с n-стиролсульфо-кислотной, α, β-ненасыщенными одно- и двухосновными карбоновыми кислотами и их солями, является мультикомпонентность процесса в ионизирующих средах. Она заключается в том, что в системе имеет место зависящее от характера среды равновесие между различными формами сосуществования положительно и отрицательно заряженных частиц:
δ- δ+
![]()
![]()
![]()
![]()
![]()
![]() А Х А- Х+ А- IIХ+ А- + Х+
А Х А- Х+ А- IIХ+ А- + Х+
Общая схема ионизационного равновесия не постулирует одновременного существования в системе всех четырех форм ионогенного мономера [молекулярной, ионной (контактные и разделенные пары) и свободных ионов], таких форм может быть три или две (например, А- IIХ+ и А- +X+ ) в зависимости от характера реакционной среды. Следствием мультикомпонентности системы является осложненный [87, 114] характер сополимеризации. Поэтому активность мономеров в реакции сополимеризации зависит от общей концентрации мономеров и состав; исходной мономерной смеси [115, 52, 116, 117], ионной силы растворов [52, 118-123], полярности растворителя [124] и степени превращение [125, 126]. При сополимеризации с ионогенными мономерами наблюдается также сильная зависимость конформационного состояние макромолекул от характера реакционной среды [52, 127, 128].
С уменьшением диэлектрической постоянной смеси воды и ДМСО начальная скорость сополимеризации АА с натриевой и калиевой солями n-стиролсульфокислоты понижается. Наблюдаемое при этой понижение реакционной способности амида связано со смещением равновесия между ассоциацией амида и его сольватацией в сторону последней, ростом комплексообразования между макрорадикалами ДМСО, уменьшением размеров макромолекулярных клубков, ведущим к понижению локальной концентрации мида в области, где имеются активные центры [52, 124].
Ввиду практической значимости сополимеров МАА и МАК целессообразно более подробное рассмотрение их синтеза. При получении этих сополимеров в 40%-х водных растворах (85 °С) по мере увеличение степени нейтрализации кислоты гидроксидом натрия (росте рН) отнссительная активность амида растет (от 0,28 до 0,64), а кислоты падает (от 2,6 до 0,4) [117]. С повышением рН уменьшается доля протонированных молекул амида и радикалов, на концах которых находятся элементарные звенья протонированного амида, и увеличивается степень диссоциации кислоты и соответствующего макрорадикала, т.е. имеют место ослабление отталкивания амидного радикала молекулы амида, усиление отталкивания кислотного радикала молекулы кислоты (анионов). Следовательно, увеличение r1 и уменьшение r2 могут быть обусловлены ростом k11 и падением k22 [117].
При сополимеризации АА и МАК наблюдается качественно такая же картина, как и при сополимеризации МАА и той же кислоты: при рН < 3, когда кислота очень слабо ионизирована, а константы скоростей роста и обрыва при ее гомополимеризации не зависят от концентрации ионов водорода, величины r1 и r2 практически постоянны при изменении рН. При этом r2 превышает r1 в еще большей степени, чем системе МАА — МАК. При рН > 3 значение r2 резко понижается [129].
Поскольку в системах амид – кислота оба компонента могут обуcлов-ливать «особый» характер систем, вполне естественно, что при сополимеризации до глубоких конверсии значения r1 и r2 недрерывно меняются. Непостоянство r1 и r2 в ходе сополимеризации АА и ненасыщенных кислот впервые было установлено при использовании малеината натрия, сукцината натрия и других солей [87, 121, 130] в качестве второго мономера.
На основании кинетических данных по сополимеризации АА и АК до 80%-й конверсии была сделана попытка определить относительной активности мономеров по методу Келена-Тюдоша [108], что, однако, не удалось (значения r1 и r2 оказались равными соответственно 0,50 ± 0,06 и 0,79 + 1,67). Колебания r2 в столь широких пределах, очевидно, обусловлены изменением реакционной способности в ходе сополимеризации, хотя сами авторы такого вывода ие делают [131].
Экспериментальные данные по кинетике начального периода сополимеризации в 7%-х (масс.) водных растворах МАА и метакрилата натрия, взятых в различных соотношениях, удовлетворительно описываются [132] известным уравнением [112, с. 377], которое предложили Мелвилл, Нобл и Уотсон. Согласно данному уравнению, обрыв контролируется химическими реакциями, а диффузионные процессы не учитываются. Вместе с тем, именно ввиду влияния диффузии на закономерности обрыва цепи указанное уравнение очень часто оказывается неприменимым к описанию кинетики сополимеризации. Предположено [132], что возможность использования уравнения при сополимеризации МАА и метакрилата натрия связана с тем, что в данной системе константы скоростей реакций обрыва (за счет взаимодействия одинаковых и различных радикалов) близки между собой [133]. В системе МАА — метакрилат натрия кривая зависимости начальной скорости сополимеризации от соотношения между мономерами проходит через слабо выраженный максимум, что при относительной близости констант скоростей обрыва определяется предпочтительностью перекрестного роста по сравнению с ростом за счет любой гомополимеризации (r1 < 1 и r2 < 1 [117]). Для системы АА – АК (вода, рН = 4,6) также наблюдается превышение скоростью сополимеризации скоростей гомополимеризации обоих мономеров [134].
Сополимеризация МАА и МАК (или ее соли) протекает без самоускорения [135]. Гель-эффект, по-видимому, перекрывается уменьшением отдельных констант скоростей роста при увеличении конверсии мономеров.
При сополимеризации в воде АА и акрилата калия в присутствии твердого инициатора, нерастворимого в реакционной смеси, образуется сополимер, содержащий меньше АА, чем сополимер, полученный в присутствии водорастворимого инициатора, что, по-видимому, связано с избирательной адсорбцией акрилата калия на твердом инициаторе [136].
Сополимеризация с ненасыщенными аминами и их солями. Представляют практический интерес катионные сополимеры АА с аллил-амином и замещенными аллиламинами. При их получении АА оказывается значительно более активным в сополимеризации, чем сомономер. Так, при сополимеризации с АА гидрохлорида алллиламина (вода; рН = 3,0, 40 °С) r1 = 13,35 ± ± 0,26 и r2 = 0,08 ± 0,02 [137], диаллил-диметиламмонийхлорида (вода; рН = = 6,1; 40 °С) r1 =6,7 и r2 = 0,58 [138]. В отличие от мономеров, содержащих фрагменты аллиламина и дающих при сополимеризации относительно стабильные радикалы, другие амин- и аммонийсодержащие сомономеры обычно превосходят по активности АА. При сополимеризации АА с 4-диметил-аминостиролом (метанол; 60 °С) r1 = 0,15 и r2 = 3,35 [139], с 5-винил-1-метил-2-пиколи-нийметилсульфатом (вода; 48 °С) r1 = 0,19 и r2 = 2,7 [140].
Весьма подробно изучена сополимеризация АА и МАА с мономерами, в молекулах которых аминогруппа отделена от винильной группы цепочками из 4 и более атомов, в первую очередь — с диалкиламино-алкил(мет)акрила-тами. При гетерофазной сополимеризации в ацетоне МАА с диалкиламиноэтилметакрилатами в виде неионизированных оснований процесс близок к идеальному, r1 и r2 мало отличаются от единицы) [141]. Такая же картина наблюдается при сополимеризации N,N-диметиламиноэтилметакрилата (ДМАЭМ) с ММА. Близость относительных активностей к единице указывает на то, что скорости роста цепей в этих системах контролируются скоростью диффузии молекул мономеров в макромолекулярные клубки, причем скорости диффузии сомономеров мало различаются между собой [142].
Переход от диалкиламиноэтилметакрилатов к их солям при сополимеризации в воде приводит к резкому изменению значений относительных активностей мономеров. Так, при сополимеризации (вода; 70 °С) МАА с гидрохлоридом ДМАЭМ r1 = 0,26 ± 0,13 и r2 = 2,6 ± 0,14, с гидрохлоридом N,N-диэтиламиноэтилметакрилата (ДЭАЭМ) – r1 =0,17 ± 0,04 и r2 = 0,39 ± 0,01 [335, 336]. Предположено, что положительные заряды макромолекулы соли способствуют выпрямлению цепи и освобождению конца макрорадикала, что делает его более доступным для молекул мономера, благодаря чему скорость роста контролируется скоростью химической реакции и зависит от строения реагирующих частиц, т. е. константы скорости элементарных реакций роста при сополимеризации, как правило, уже не могут быть равны между собой [142]. Уменьшение r1 и увеличение в отдельных случаях r2 при переходе от свободных оснований к их солям связано с тем, что амиды в целом менее реакционноспособны при взаимодействии со свободными радикалами, чем стерически затрудненные соли на основе N,N-диалкиламиноэтилметакрилатов. Это может быть обусловлено образованием в молекулах солей замкнутых систем (за счет притяжения между аммонийным атомом азота и карбонильным атомом кислорода), способствующих делокализации неспаренного электрона на а-углеродном атоме и, тем самым, относительно большей стабильности; соответствующих радикалов, чем амидных радикалов, в результате чего k11 < k12 и k22 > k21. Вместе с тем, значение r2 < 1 в системах МАА – соль на основе ДЭАЭМ указывает, что рассматриваемые константы сополимеризации зависят и от других факторов. Одним из них может быть электростатическое отталкивание между одноименно заряженными молекулой и радикалом солеобразного производного ДЭАЭМ [141].
Значения r1 и r2 для α, β-ненасыщенных амидов с ДЭАЭМ или его солеобразными производными оказались независящими от степени превращения мономеров при сополимеризации [143, 144], в то время как при сополимеризации с кислотами или нитрилами они в ходе процесса резко изменяются. Данное различие, вероятно, объясняется тем, что диалкиламиноалкил(мет)акрилатные звенья вследствие наличия в них занимающих сравнительно большой объем диалкиламиноалкильных остатков в условиях сополимеризации стерически препятствуют ассоциации мономерного амида с амидной группой в сополимере [143].
Сополимеризация амидов с солеобразными производными диалкил-аминоалкил(мет)акрилатов протекает со значительно большей скоростью и приводит к получению более высокомолекулярных сополимеров, чем при сополимеризации со свободными основаниями. Это можно объяснить меньшей (из-за электростатического отталкивания) скоростью реакций обрыва, в которых участвуют два макрокатионрадикала, чем реакции обрыва, основанной на столкновении незаряженных частиц, а также имеющим место при переходе от свободных оснований к солям развертыванием растущих макроцепей и освобождением реакционных центров, способствующим реакции роста при сополимеризации. Вместе с тем, сополимеризация амидов с диал-киламиноалкилметакрилатами в присутствии двухкратного избытка НСl по отношению к аминам не дает заметного эффекта по сравнению с сополимеризацией в отсутствие НСl. Благодаря экранированию положительных зарядов избытком противоионов хлора растущие цепи свернуты в клубки и подход к ним молекул мономера так же стерически затруднен, как и при сополимеризации со свободными основаниями. Таким образом, для получения сополимеров амидов с диалкиламиноалкил(мет)акрилатами с большой скоростью и достаточной вязкостью следует предварительно нейтрализовать основание, либо перевести его в четвертичную аммониевую соль. Аналогичный результат достигается путем совмещения процессов алкилирования диалкиламиноалкил(мет)акрилата и сополимеризации его с амидом [141-143].
Сополимеризацию с солями на основе диалкиламиноалкил(мет)-акрилатов проводят в присутствии пероксидных инициаторов, с диалкиламиноалкил(мет)акрилатами в виде свободных оснований – в присутствии инициаторов, не взаимодействующих с аминогруппой (азосоединений). Сополимеризация МАА и не нейтрализованных диалкил-метакрилатов в ацетоне практически прекращается по достижении 60-70%-й конверсии мономеров, несмотря на наличие инициатора [142].
В работе [145] при сополимеризации АА и МАА с гидрохлоридом ДЭАЭМ (мольное соотношение 4:1) в водных растворах до глубоких степеней превращения получены сополимеры, плохо растворимые в воде. В обеих системах ввиду протекания процесса в кислой среде возможно сшивание макромолекул за счет образования межмолекулярных вторично-амидных (-CONHCO-) мостиков. Кроме того, в случае системы на основе ММА, вследствие более высоких значений r2 по сравнению с r1, при больших конверсиях образуются плохо растворимые в воде фракции, обогащенные звеньями амида. Данное объяснение согласуется с тем, что улучшить растворимость сополимера МАА и гидрохлорида оказалось возможным путем дозирования при сополимеризации более активного мономера – гидрохлорида ДЭАЭМ. При этом одновременно повышалась степень однородности по составу макромолекул сополимера.
Глава II. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
2.1. Подготовка исходных реагентов и растворителей
Ацетон – сушили над CaCl2, а затем кипятили над оксидом фосфора (Р2 О5 ) в течение двух часов и дважды перегоняли над Р2 О5 , Ткип = 56 °С, n![]() = 1,3550.
= 1,3550.
Этанол абсолютный. Этанол (ректификат) восемь часов кипятили над свежепрокаленным оксидом кальция (на 1 литр этанола 200г СаО), затем смесь 5г магниевых стружек, 0.5г сублимированного йода и 75 мл спирта, высушенного над СаО, кипятили с обратным холодильником до полного превращения Мg в этилат магния. В реакционную колбу добавляли остальное количество спирта, раннее обработанного СаО, кипятили смесь в течение 2-х часов и отгоняли спирт.
Акриловую и метакриловую кислоты марки «ч» (чистый) сушили над CaCl2 и перегоняли над рафинированными медными стружками под вакуумом, для АК: Ткип = 26-28 °С (3-5 мм. рт. ст.), d20 = 1,0511, n![]() = 1,4224; для МАК: Ткип = 56-57 °С (10 мм. рт. ст.), d20 = 1,015, n
= 1,4224; для МАК: Ткип = 56-57 °С (10 мм. рт. ст.), d20 = 1,015, n![]() =1,4315. Тпл =13,5 °С для АК и Тпл =16,0 °С для МАК.
=1,4315. Тпл =13,5 °С для АК и Тпл =16,0 °С для МАК.
Диэтиловый эфир сушили над щелочьюи дважды перегоняли над металлическим натрием. Отогнанный эфир хранили над натрием, Ткип. = 34-35 °С.
Акриламид – CH2 =CH–CONH2 «ч.» фирмы «Acros», Тпл = 84,5 °С.
Пропанол – сушили над CaО и перегоняли.
Метанол марки «х.ч.» (химический чистый), Ткип. = 64,5-65,0 °С.
Гидроксид натрия, хлорид натрия, серная кислота марки «х.ч.»
Гидрохлорид гуанидина (ГГХ) «х.ч».
Инертный газ-аргон (о.с.ч). (особо чистый)
Во всех опытах использована бидистиллированная вода.
Очистка инициатора. Персульфат аммония (ПСА) (NH4 )2 S2 O8 ,µ-пероксо-бис(тетраоксосульфат) аммония, «ч.д.а.» (чистый для анализа), перекристаллизовывали из бидистиллированной воды несколько раз, затем высушивали в вакууме до постоянного веса.
2.2 Синтез мономерной соли на основе метакриловой кислоты и гуанидина
а) Получение этилата натрия.
В трехгорлую колбу (объемом 2 л) снабженную мешалкой и обратным холодильником, помещали необходимое количество абсолютированного этилового спирта (500 мл) и при непрерывном перемешивании понемногу добавляли металлический натрий (1 моль) в течение двух часов. После добавления всего натрия, реакционную массу перемешивали еще 1 час до полного растворения натрия.
б) Получение гуанидина.
В полученный раствор этилата натрия при перемешивании порциями добавляли эквимольное количество (1 моль) ГГХ. Реакционный раствор перемешивали в течение 4 часов и оставляли в холодильнике на ночь. На следующий день раствор гуанидина отфильтровывали от выпавшего осадка хлорида натрия.
в) Получение акрилата (метакрилата) гуанидина (МАГ).
Полученный накануне реакционный раствор гуанидина помещали в колбу, снабженную мешалкой, термометром, капельной воронкой. После охлаждения раствора гуанидина до – 10 – 6 °С при перемешивании прикапывали акриловую (метакриловую) кислоту в течение двух часов. Скорость подачи регулировали так, чтобы температура в реакционной массе не превышала -5 – 0 °С. Для отвода избыточного тепла применяли охлаждающую баню из смеси ацетона и сухого льда. После прикапывания всего количества метакриловой кислоты, рН реакционного раствора был близок к 7. Раствор перемешивали еще 4 часа при комнатной температуре, после чего колбу с продуктом реакции помещали в холодильник на несколько суток.
Полученный прозрачный раствор мономерной соли в этаноле высаживали в 10-кратный избыток абсолютированного эфира. Выпавшие белые игольчатые кристаллы АГ или МАГ отфильтровывали через плотный стеклянный фильтр, многократно промывали абсолютным эфиром и сушили в вакууме при комнатной температуре. Выход АГ – 70-75 %, МАГ – 80-85 %. Синтезированную соль подвергали двукратной перекристаллизации из смеси абсолютного спирта и ацетона 10:90.
Структура и состав всех синтезированных мономерных солей была подтверждена совокупностью различных физико-химических методов анализа: ЯМР и ИК-спектроскопией, элементным анализом, Т.пл.
2.3 Кинетические измерения
Кинетика полимеризации мономера АГ, а также их сополимеризация с ММГ измерялась методом дилатометрии. Приготовленная реакционная смесь трижды дегазировалась на вакуумной установке (10–3 мм рт. ст.) в специальных ампулах, после чего раствор в вакууме переливался в дилатометр, и система продувалась аргоном особой чистоты. Дилатометр снимался с установки, закрывался стеклянной пробкой и помещался в термостат (Т =60°С). После термостатирования уровень раствора в капилляре дилатометра доводили до требуемого, отбирая часть его шприцом с длинной иглой. Скорость полимеризации определяли по данным об изменении объема реакционного раствора, регистрируя изменение уровня раствора в капилляре дилатометра с помощью катетометра КМ-6 (точность измерения ± 0,001 см).
Степень превращения мономера в полимер определяли исходя из значений контракции, определенной методом пикнометрии, которая для реакции полимеризации АГ в воде равнялась 10,8 %, МАГ – 7 %, АА – 14%.
Начальные скорости полимеризации находили по тангенсу угла наклона начальных линейных участков соответствующих кривых зависимости степени превращения от продолжительности полимеризации. Относительная ошибка не превышает 5 %.
2.4 Выделение и очистка полимеров из реакционных растворов
Сополимеры из реакционной массы выделяли диализом относительно дистиллированной воды с использованием диализных мешочков фирмы «Spectrapor» (США). Затем свободные от мономеров растворы переносились в кристаллизаторы, вода испарялась (при комнатной температуре), оставшийся полимер сушили в вакуумном сушильном шкафу над P2 O5 при 40-50 °С (~ 40-50 часов) либо из водного раствора высаживали в ацетон, затем фильтровали и сушили.
2.5 Вискозиметрические измерения
Вискозиметрические измерения исходных мономерных растворов проводили в вискозиметре Убеллоде при 30 °С. Характеристические вязкости полимеров определяли методом вискозиметрии в 1н растворе NaCl в воде при 30 °С.
2.6 Физико-химические методы исследования
Спектры ЯМР 1 Н измеряли на спектрометре “Bruker MDS-300” (300мгц) в D2 О, Ме2 СО-d6, СD3 OD и DMSO-d6 при 20 °С, химические сдвиги определены относительно остаточных протонов растворителя.
ИК-спектры синтезированных мономеров и полимеров записывались на спектрофотометре “Spekord M82” (Carlzeis Jena). Образцы для снятия спектров готовились прессованием таблеток смеси исследуемого образца с бромидом калия (1:200), тщательно растертой в агатовой ступке. Давление пресса составляло 4-6 т/см2. Полученные таким образом таблетки не растрескивались и оставались прозрачными в течение нескольких часов.
Термофизические методы исследования. Фазовое поведение систем изучали на термоаналитической установке (МОМ, Венгрия) с нагревательной ячейкой DSС-30 при скорости нагревания 5 град/мин в саморегенерирующейся атмосфере воздуха.
Термическую устойчивость изучали методом дифференциального термического анализа (ДТА) и дифференциальной термической гравиметрии (ДТГ) на дериватографе Q-1000D (фирма МОМ, Венгрия) на воздухе при скорости нагревания 2,5 град/минот 20 до 1000 °С. Термофизические данные обрабатывались с использованием программно-аппаратного комплекса для измерения сигналов дериватографа Q-1000D (фирма МОМ, Венгрия) и компьютерной обработки данных термогравиметрического анализа.
2.7 Фотометрический метод определения мутности
Мутность воды определяли фотометрическим путем сравнения проб исследуемой воды со стандартными суспензиями на КФК-3.
Построение градуировочного графика
Градуировочный график строили по стандартным рабочим суспензиям.
![]()
Рис. 5. Калибровочный график определения мутности воды
Перед проведением испытания во избежание ошибок производили калибровку фотоколориметра по жидким стандартным суспензиям мутности или по набору твердых стандартных суспензий мутности с известной оптической плотностью.
Содержание мутности в мг/л определяли по градуировочному графику рис 5.
Определение параметров флоккулирующей активности сополимеров
Флоккулирующую активность полиэлектролитов определяли на модельных и реальных дисперсных системах. Одной из модельных систем являлась водная суспензия каолина, которая в поверхностном слое частиц имеет отрицательный заряд за счет силанольных групп. Каолин представляет собой вид белой глины, состоящий из оксидов кремния (30-70%) и алюминия (10-40%). Главная составная часть каолина — минерал каолинит (подкласс слоистых силикатов) Al4 [Si4 O10 ](OH)8, плотность 2500-2700 кг/м3 .
Каолин сушили до постоянного веса при 100 °С и хранили в эксикаторе. Суспензию готовили за 1 сутки до проведения измерений, концентрация дисперсной фазы в суспензии составляла 0,8 % (масс). Перед проведением анализа суспензию перемешивали с целью равномерного распределения частиц.
Для исследования процесса флоккуляции в водную суспензию каолина с концентрацией 0,5 и 0,8 % вводили флоккулянты, концентрация которых в системе составляла 0,01-1,0 масс. %, и определяли зависимость мутности надосадочной жидкости от концентрации и состава сополимера.
При проведении турбидиметрических измерений полученные полимерсодержащие суспензии перемешивали в течение 2-х мин со скоростью 100 об/мин, не допуская попадания воздуха. Через определенные промежутки времени отбирали надосадочную жидкость и измеряли оптическую плотность (D) на приборе КФК-3 при длине волны λ = 364 нм против дистиллированной воды.
2.8 Определение бактерицидных свойств сополимеров
Методика 1. Для определения числа колоний кишечной палочки производят посев исследуемой воды, обработанной гуанидиновыми препаратами, на мясо-пептонную среду и выращивают колонии E.coli в течение 48 часов при 22 ºС, после чего колонии пересчитываются. Таким образом определяются бактерицидные свойства гуанидинсодержащих полимеров непосредственно в воде.
Порядок анализа. Расплавляют в баке с теплой водой (35ºС) стерильную питательную среду.
Ставят на стол в один ряд 4 стерилизованных чашки Петри и, слегка приподнимая только один край крышки, наливают в них при помощи стерилизованной пипетки по порядку: 0,1 – 0,2 – 0,5 – 1,0 см3 исследуемой воды. Затем в каждую из чашек прибавляют около 10 см3 питательной среды и плавными движениями смешивают ее с водой, равномерно распределяя массу по дну чашки Петри и не пачкая ее краев. Затем чашки ставят на холодную горизонтальную поверхность и дают желатиновой среде застыть. При посеве концы пипеток должны быть фламбированы.
Чашки с застывшей средой помещают на 48 часов в термостат при температуре 20-22 ºС. По истечении этого времени производят подсчет выросших колоний. Для облегчения подсчета рекомендуется применять разграфленную на квадратные сантиметры пластинку, на которую ставят чашку Петри с колониями.
Если число выросших колоний небольшое (не превышает 300), их сосчитывают полностью все. Если выросло очень много колоний, то сосчитывают их только на 20-30 отдельных квадратах, расположенных равномерно по всей площади чашки Петри. Полученные цифры колоний складывают вместе, делят на число подсчитанных квадратиков и таким образом определяют среднее число их на 1 см3 . Подсчет колоний производят с лупой (увеличение 6-8 раз).
Затем вычисляют площадь чашки Петри в квадратных сантиметрах и, умножив полученную величину на среднее число колоний на 1 см2 , находят их общее число в том количестве воды, которое было взято для посева в данной чашке.
Методика 2. Для количественной оценки биоцидного действия гуанидинсодержащих полимеров пользовались методом диффузии в агар. Для проведения теста с диффузией чашки заполняли до определенной высоты агаризированной средой, содержащей суспензию тест-организма (E.coli). Затем в чашки вносили минимальные количества исследуемого полимера (1 мг для гуанидинсодержащих полимеров). Это количество вносили в лунки питательной среды. При положительной реакции во всех случаях после инкубации становится заметной зона подавления роста тест-организма (рис. 6). Диаметр этой зоны при соблюдении постоянных условий опыта (состав питательной среды, толщина слоя агара, плотность посева, время инкубации, температура и т. д.) пропорционален логарифму концентрации биоцидного полимера.
Глава III . ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
3.1 Радикальная сополимеризация акрилат и метакрилатгуанидинов акриламидом в водных средах
Несмотря на важные мирные «профессии» полимеров АА, их использование в оборонной промышленности значительно ограничило доступность научной информации, поэтому до начала 70-х годов в литературе отсутствовали сведения о технологии производства указанных полимеров. В последние годы наряду с улучшением сырьевой базы создана научная основа для направленной разработки полимеров с заданными свойствами, разработаны перспективные методы синтеза полимеров – полимеризация и сополимеризация АА в концентрированных водных растворах и дисперсиях, получили развитие методы химической модификации полимеров. Сополимеры АА традиционно и широко используется во многих отраслях промышленности, в том числе, в качестве флоккулянтов [146-151], в нитроцеллюлозном, керамическом и силикатном производстве [153], при очистке сточных вод [146-155], для осаждения полимерных латексов [154-155], стабилизации взвесей и пищевых жидкостей [153].
В сельском хозяйстве они, а также полимерные комплексы на их основе, служат для улучшения структуры почв, для предотвращения ветровой эрозии [154], и данный перечень применений далеко не полный и составляет лишь небольшую часть хорошо известных и широко используемых полимерных материалов на основе сополимеров АА.
В настоящее время полимеры АА производят крупные фирмы США, Японии и развитых стран Европы. Они являются основными поставщиками полимеров на мировой рынок, а в России, Китае и ЮАР полимеры производят для внутреннего потребления. Производство полимеров АА продолжает неуклонно возрастать и к концу века достигнет 400 тыс. т. в год. Однако темпы роста производства не удовлетворяют потребностей, которые ежегодно возрастают на 8-10%. Поэтому актуальны разработка новых и совершенствование существующих перспективных методов синтеза ПАА, его производных и сополимеров АА.
Дальнейшее развитие исследований в этой области как в теоретическом, так и в практическом аспекте, несомненно, приведет к созданию новых и совершенствованию существующих перспективных методов синтеза полимеров – полимеризации и сополимеризации АА в концентрированных водных растворах и дисперсиях, развитию методов химической модификации ПАА, а также расширению сферы применения полимеров АА. В конечном итоге это будет способствовать удовлетворению растущих потребностей различных областей техники и технологии в интересных и нужных полимерах.
С учетом высокой биоцидной активности гуанидинсодержащих соединений, давно и успешно применяемых в медицине и в разных областях промышленности, представлялось необходимым изучить возможность синтеза новых сополимеров на основе гуанидинсодержащих мономеров акрилового ряда и АА. Поскольку естественно было бы ожидать, что вновь созданные сополимеры могут проявлять новые важные свойства и характеристики, не присущие исходным гомополимерам.
Вместе с ожидаемой практической значимостью указанных полимеров изучение кинетических особенностей протекания реакции радикальной сополимеризации, несомненно, актуально и в научном аспекте, прежде всего с позиции оценки реакционной способности синтезированных мономеров в рассматриваемых условиях.
Полученная в результате таких исследований информация необходима также для эффективного управления процессами получения сополимеров с заданным составом; распределением в них химических звеньев и молекулярно-массовыми характеристиками.
До проведения систематических кинетических исследований в рассматриваемых нами сополимеризационных системах были определены оптимальные условия осуществления данных реакций – водная среда; суммарная концентрация сополимеров [М] = 2 моль л–1; [ПСА] = 5×10–3 моль л–1; 60 °С.
Реакцию сополимеризации проводили по схеме 6 :

Изучение реакции сополимеризации в данных условиях показало, что реакционные растворы были гомогенны во всем интервале составов, а образующиеся сополимеры хорошо растворялись в воде.
Как известно [156], при гополимеризации АГ и МАГ наблюдается микрогетерогенность реакционного раствора при степенях превращения более 5%. Особенно, данное явление выражено для МАГ. Авторы [156] объясняют обнаруженную при полимеризации МАГ в Н2 О гетерогенность реакционной среды конформационными превращениями ПМАГ, проявляющимися в сворачивании цепи — аналогично хорошо известным процессам денатурации ряда белков, а также синтетических полимеров – аналогов белка (например, поли N-винилпирролидона, ПВП), о чем подробно сообщалось в ряде работ [157-161]. Интересно, что для ПВП, как следует из этих работ, эффективным денатурирующим агентом являются низкомолекулярные соли гуанидина. Авторы полагают, что именно наличие двух аминогрупп в молекуле гуанидина, способных конкурировать с карбонильной группой С=О, блокируя дальнейшее взаимодействие ее с молекулами растворителя (вода), вызывает резкое сворачивание цепи ПВП. Так, в присутствии гуанидингидрохлорида характеристическая вязкость ПВП в спиртовых растворах заметно падает. Особенно резко меняется К[h]2, т.е. величина, характеризующая взаимодействие между полимером и молекулами растворителя, при этом молекулы ПВП, почти полностью растворимые в спирте, становятся нерастворимыми в присутствии гуанидингидрохлорида, что является следствием блокирования кислорода пирролидонового цикла молекулами гуанидинхлорида, приводящего к увеличению сил межмолекулярной ассоциации колец ПВП посредством гидрофобных взаимодействий. Моравец и другие авторы [157-159], подробно изучавшие влияние различных факторов на денатурацию белка, установили что различные соли гуанидина оказывают сильный денатурирующий эффект на белковые молекулы при введении их в раствор даже в небольших концентрациях ~1% (см. рис. 7).


Рис. 7. Изменение формы клубка ПАГ и ПМАГ в присутствии собственного мономера или гуанидингидрохлорида
Весьма примечательно, исходя из вышесказанного, что при сополимеризации МАГ с АА удается нивелировать «денатурирующее» действие гуанидинсодержащего мономера МАГ реакция сополимеризации до высоких степеней превращения (~60%) протекает в гомогенных условиях.
Это означает, что, как и в случае природных белковых молекул, введение звеньев «чужого» «нейтрального» мономера в сополимер (каким в нашем случае является АА), приводит к нарушению тактичности (изомерного состава) полимерной цепи, и чем больше количество таких “включений” в цепь ПМАГ, тем менее выражено влияние гуанидинсодержащего мономера на гетерогенность процесса полимеризации МАГ.
Таблица 8
Скорости сополимеризации АА с МАГ в водных растворах (рН » 7) а
| № п/п | Исходный состав сополимера АА-МАГ | [M], моль л-1 | Инициатор, 5´10-3 моль×л-1 | Vр ´105 , моль×л-1 ×с-1 | Микро-гетеро-генность |
| 1 | 50:50 | 1,0 | ПСА | 1,0 | – |
| 2 | 50:50 | 2,0 | ПСА | 26,9 | – |
| 3 | 50:50 | 0,5 | ПСА | 0,36 | – |
| 4 | 70:30 | 2,0 | ПСА | 5,7 | – |
| 5 | 70:30 | 1,0 | ПСА | 0,73 | – |
Состав сополимеров АА: АГ определяли по данным элементного анализа так как химические сдвиги протонов –СН2 -СН= в ЯМР 1 Н спектрах сомономеров близки и перекрываются.
Таблица 9
Данные элементного состава сополимеров АА: АГ
Исх. состав АГ: АА | %С ± 0,20 | %N ± 0,40 | %H ± 0,20 | в сополимере |
| 80:20 | 38,85 | 29,33 | 6,90 | 0,755 |
| 50:50 | 41,99 | 26,62 | 6,96 | 0,634 |
| 40:60 | 41,85 | 26,74 | 6,80 | 0,639 |
| 20:80 | 44,15 | 24,77 | 7,30 | 0,561 |
| 10:90 | 47,37 | 22,31 | 7,00 | 0,471 |
Для расчета содержания сомономеров использовали соотношение содержания азота и углерода в сополимере R = %N/%C, исходя из соображения, что
NСП = NАГ ×x + NАА ×(1 – x), (1)
CСП = CАГ ×x + CАА ×(1 – x), (2)
где NАГ и CАГ – содержание в АГ; NАА и CАА – содержание в АА; x – доля АГ в сополимере и (1 – x) – доля АА в сополимере.
Отсюда имеем уравнение:
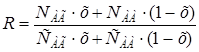 . (3)
. (3)
Решая это уравнение и подставив значения для содержания азота и углерода в соответствующих сомономерах, получаем выражения для расчета х, т.е. доли АГ в сополимере.
Расчет состава сополимеров АА с МАГ проводили по данным ЯМР 1 Н спектроскопии, используя интегральную интенсивность сигнала метильной группы сомономера МАГ, который проявляется в самом сильном поле и не перекрывается никакими другими сигналами. Треть его интегральной интенсивности будет равна величине условного протона для звена МАГ – «1Н (М2 )». Протоны, относящиеся к сигналам CH2 -групп цепи сополимера, проявляются для обоих сомономеров вместе в области химических сдвигов 1,5-1,8, поэтому для определения условного протона звена АА «1Н (М1 )» из общей интегральной интенсивности этих протонов (I) вычитали вклад двух протонов звена МАГ и оставшуюся величину делили на 2 (уравнение (4)):
![]() . (4)
. (4)
Из полученных результатов определяли мольное содержание сомономеров в сополимере, выраженное в мол.% (уравнения 5 и 6):
МПААм = [«1Н (М1 )»: («1Н (М1 )» + «1Н (М2 )»)]×100% (5)
МПМАГ = [«1Н (М2 )»: («1Н (М1 )» + «1Н (М2 )»)]×100% (6)
Как видно по кривым на рис. 8, при всех исходных мольных соотношениях сомономеров, сополимер обогащен звеньями акрилатного сомономера, причем, системе МАГ–АА свойственно большее обогащение сомономером МАГ, в отличие от системы АГ–АА. Это свидетельствует о большей реакционной способности МАГ в реакции радикальной сополимеризации и соответствует данным о параметрах реакционной способности акриловой (АК) и метакриловой (МАК) кислоты, имеющимися в литературе. Большая в сравнении с АГ реакционноспособность мономера МАГ обусловлена, возможно, большей делокализацией заряда карбоксильной группы в молекуле мономера, на что указывает смещение сигналов винильных протонов МАГ в более сильное поле по сравнению с АГ в ЯМР1 Н спектрах.
![]()
Рис. 8. Зависимость состава образующихся сополимеров в системах:
АГ-АА (кривая 1) и МАГ-АА (кривая 2)
от состава исходного реакционного раствора
Меньшая реакционная способность акриламида по сравнению с АГ и МАГ может быть обусловлена со специфическим строением ионогенных мономеров, в которой имеется электростатическое притяжение между положительно заряженным атомом аммонийного азота и карбонильным атомом кислорода остатка метакриловой кислоты, электронная плотность у которого повышена (схема 7).
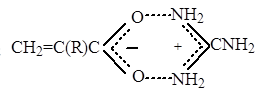
где R= H, СН3
Схема 7. Цвиттер-ионная делокализованная структура АГ и МАГ
Это притяжение обуславливает делокализацию отрицательного заряда по связям карбоксилат-аниона акриловой и метакриловой кислоты. Вследствие такой делокализации относительная стабильность соответствующих радикалов выше по сравнению с акриламидом. В случае МАГ наблюдается более высокая делокализация электронов по связи С-О– в метакрилатанионе по сравнению с АГ, что подтверждается большим обогащением сополимеров сомономером МАГ по сравнению с АГ.
Для определения констант сополимеризации в бинарной системе на практике используются различные методы, в основе которых лежит уравнение состава сополимера (7) [162]:
 , (7)
, (7)
где [M1 ] и [M2 ] — концентрации мономеров в исходной смеси; r1 и r2 — константы сополимеризации, r1 =k11 /k12 и r2 =k22 /k21 .
Одни методы могут применяться только к низким конверсиям мономера (до 8%), в них делается допущение, что на начальной стадии сополимеризации сохраняется постоянство величин М1 и М2. Поэтому соотношение скоростей расходования мономеров можно заменить соотношением мольных концентраций мономерных звеньев [m1 ] и [m2 ] в сополимере:
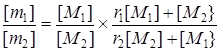 . (8)
. (8)
Это, например, метод «пересечения прямых» Майо-Льюиса [163], аналитический метод вычисления констант сополимеризации [164] и др.
Разработаны методы расчета констант сополимеризации, которые позволяют определять состав мономерной смеси или сополимера практически при любой конверсии мономеров, т.к. уравнения состава решаются в интегральной форме. Наиболее простым из них является метод Файнемана-Росса [165].
Так как нами исследовалась сополимеризация на малых степенях конверсии, то для расчета констант сополимеризации мы использовали аналитический метод.
Основное уравнение аналитического метода, предложенного А.И.Езриелевым, Е.Л.Брохиной и Е.С.Роскиным [210] имеет следующий вид:
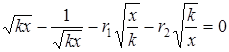 , (9)
, (9)
где x = [M1 ]/[M2 ]; k = [m1 ][M1 ]/[m2 ][M2 ], а [mi ] и [Mi ] – концентрации i-ого компонента в полимере и исходной мономерной смеси. Уравнение (9) уже симметрично относительно величин r1 и r2, поэтому обе константы определяются с одинаковой точностью.
Это уравнение также удобно для вычисления констант сополимеризации методом наименьших квадратов (МНК). В последнем случае соответствующие уравнения имеют вид:
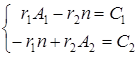 ,
,
где
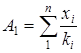 ;
; 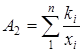 ;
; 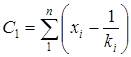 ;
; 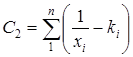 ,
,
а n – число опытов.
Тогда выражение для относительных активностей мономеров записывается как:
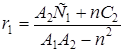 и
и 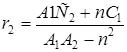
Аналитический метод позволяет рассчитать среднеквадратичную ошибку определения констант сополимеризации
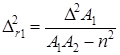 ;
; 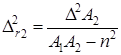 ,
,
где 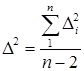 дает среднеквадратичную ошибку опыта, т.е.
дает среднеквадратичную ошибку опыта, т.е.
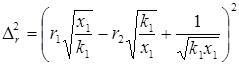
Значения констант, рассчитанные этим методом, представлены в табл. 10.
Так как нами исследовалась сополимеризация на малых степенях конверсии, то для расчета констант сополимеризации использовали аналитический метод и значения констант, рассчитанные этим методом, представлены в табл. 10.
Таблица 10
Значение эффективных констант сополимеризации в системах
АГ(МАГ) (М1 ) –АА (М2 )
([М]сум = 2 моль×л–1; [ПСА] = 5×10–3 моль×л–1; 60 °С, Н2 О)
| № пп | Сополимеризационная система | r1 | r2 | r1´r2 |
| 1 | АГ-АА | 0,73 ± 0,07 | 0,27 ± 0,01 | 0,197 |
| 2 | МАГ-АА | 0,94 ± 0,05 | 0,77 ± 0,04 | 0,723 |
Приведенные в табл. 10 значения r1 < 1 и r2 < 1 свидетельствуют о предпочтительном взаимодействии макрорадикалов с «чужим», чем со «своим» мономером в обеих сополимеризационных системах. Значения произведения r1 ×r2 < 1 говорит о выраженной тенденции к чередованию в обеих сополимеризационных системах. Кроме того, r1 > r2, что подтверждает, что вероятность присоединения радикалов сомономеров к мономерной молекуле МАГ и АГ несколько выше, чем к молекуле АА. Близость относительных активностей к единице при сополимеризации МАГ–АА указывает на то, что скорости роста цепей в этой системе контролируется скоростью диффузии молекул мономеров в макромолекулярные клубки, причем скорости диффузии сомономеров мало отличаются между собой.
Таким образом, радикальная сополимеризация АА с АГ и МАГ позволяет получать сополимеры с высоким содержанием ионогенных групп.
Однако несмотря на то, что полученные нами значения относительных активностей указывают на более низкую реакционную способность мономера АА по сравнению с МАГ и АГ, изучение сополимеризации указанных сомономеров в водных растворах показало, что по мере увеличения концентрации ионогенных сомономеров АГ и МАГ в исходной реакционной значения характеристической вязкости снижаются.
Для понимания механизма сополимеризации АГ и МАГ с АА исследовали скорость данного процесса в водном растворе дилатометрическим методом. Для инициирования использовали персульфат аммония (ПСА).
Изучение кинетики в данных условиях показало, что реакция сополимеризации АГ и МАГ с АА протекает только в присутствии радикальных инициаторов и полностью подавляется при введении в реакционный раствор эффективного радикального ингибитора 2,2,6,6-тетраметил-4-оксилпиридил-1-оксила. Спонтанная реакция – полимеризация в отсутствии радикального инициатора – также не наблюдается.
Реакционные растворы были гомогенны во всем интервале составов, а образующиеся сополимеры хорошо растворялись в воде.
Показано, что в изучаемой реакции зависимость степени конверсии от продолжительности реакции в выбранных условиях (водная среда; суммарная концентрация сополимеров [М] = 2×моль×л–1; [ПСА] = 5×10–3 моль×л–1; 60 °С) характеризуется линейным участком кинетической кривой до конверсии 5-8 % .
Изучение кинетики сополимеризации показало, что с увеличением содержания ионогенного мономера в исходной мономерной смеси значения начальной скорости полимеризации v0и [h] симбатно уменьшаются при сополимеризации АА с АГ и МАГ, причем для первой системы (при полимеризации с АГ) ход данной зависимости выражен более резко. Полученные результаты хорошо согласуются с известными данными, полученными в работах [6, 7] при изучении кинетики сополимеризации ДАДМАХ с АК и МАК в водных растворах. В этих системах установлено также, что скорость сополимеризации уменьшается с увеличением содержания ДАДМАХ в исходном реакционном растворе, причем для АК это увеличение выражено в большей степени, чем для МАК.
![]()
Рис.9. Зависимость начальной скорости сополимеризации (1,4) и характеристической вязкости (2,3) сополимера МАГ с АА (1,2) и АГ с АА (3,4) от содержания ионогенного мономера в исходной реакционной смеси.
Из рис. 9 следует также, что наиболее высокомолекулярные образцы сополимеров (суждение по значениям [h]) получаются в мономерных смесях, обогащенных АА.
Наиболее вероятная причина наблюдаемого уменьшения константы скорости роста цепей с увеличением концентрации ионогенного сомономера заключается в том, что концентрация сильно гидратированных акрилат- и метакрилатанионов в относительно гидрофобных незаряженных клубках макрорадикалов оказывается ниже их средней концентрации в растворе, косвенным подтверждением чему является снижение приведенной вязкости раствора сополимера с увеличением содержания звеньев АГ и МАГ.
Уменьшение [h] логичнее связать со структурирующим действием ионов АГ и МАГ на молекулы воды, которое приводит к уменьшению объемных эффектов, т.е. качество воды как растворителя для ПААм ухудшается.
Очевидно, что явления, наблюдаемые при радикальной сополимеризации с участием ионизующихся мономеров АГ и МАГ, не могут быть объяснены только на основании классических представлений и параметры r1 и r2 могут служить лишь в качестве условных величин, отражающих влияние тех или иных факторов на поведение данного мономера при сополимеризации.
Таким образом, наблюдаемые особенности и различия в ряду рассматриваемых мономеров объясняются сложным характером вкладов различных физико-химических процессов, определяющих протекание реакции сополимеризации акриламида с гуанидинсодержащими мономерами акрилового ряда. Вместе с тем, основной вклад в изменение эффективной реакционноспособности полимеризующихся частиц вносят, ассоциативные взаимодействия между гуанидиновыми и карбоксильными группами (как внутри- так и межмолекулярные) и структурная организация соответствующих мономеров и полимеров в процессе сополимеризации.
Для установления уравнения общей скорости сополимеризации АА с АГ и МАГ проводили опыты для переменных концентраций АА, АГ, МАГ и компонентов инициирующей системы при сохранении постоянства концентраций остальных компонентов реакционной системы и условий реакции.
3.2 Радикальная сополимеризация мономалеината гуанидина с акрилат- и меткрилатгуанидином в водных средах
Ионообменные сорбенты, коагулянты и флоккулянты, биоциды, разделительные мембраны, структураторы почв, модели биополимеров, полимерные носители различного рода функциональных фрагментов – таков далеко не полный перечень практического применения синтетических полиэлектролитов. Один и распространенных и перспективных путей получения полиэлектролитов – радикальная полимеризация и сополимеризация мономеров, ионизующихся в водных растворах.
В настоящей работе рассматривается синтез биоцидного сополимера на основе акрилат- и метакрилатгуанидина с мономалеинатом гуанидина. Радикальная гомополимеризация и сополимеризация гуанидинсодержащих соединений является объектом исследования многих авторов [166-170], главным образом в связи с возможностью получения полимерных материалов с комплексом специфических свойств, в том числе и биоцидных. Однако в литературе мало сведений относительно изучения процессов радикальной сополимеризации ионогенных мономеров, содержащих одинаковые функциональные группы. В связи с этим, изучение процессов сополимеризации гуанидинсодержащих ионогенных мономеров представляется нам весьма актуальным. Известно [170], что малеинаты в силу симметричности строения, пространственных факторов и высокой положительной полярности винильной группы не образует гомополимеров в присутствии радикальных инициаторов. Экспериментальные результаты, полученные в данной работе также показали, что гомополимеризация мономалеината гуанидина (ММГ) в исследованных условиях затруднена. Так, например, степень превращения мономера ММГ в полимер в условиях ([ММГ] = 2 моль×л–1; 60 °С; [ПСА] = 5×10–3 моль×л–1 ;H2 O; время полимеризации 72 часа) составляет около 3% ([η] = 0,03 дл×г–1 ). Все эти факты говорят о существенном вкладе указанных выше факторов в процесс гомополимеризации исследованной нами системы.
Вместе с тем, существенно отметить, что при исследовании реакции радикальной сополимеризации ММГ с метакрилатом гуанидина (МАГ) был получен ряд сополимеров различного состава с достаточно высокими характеристическими вязкостями, и, следовательно, молекулярными массами.
Радикальную сополимеризацию исследовали в водных (бидистиллят), водно-метанольных и метанольных растворах, в качестве инициаторов использовали радикальные инициаторы персульфат аммония (ПСА) и динитрил азобисизомасляной кислоты (ДАК) ([I] = 10–2 -10–3 моль×л–1 ) в интервале температур 20 – 60 °С.
Предварительно было установлено, что в отсутствии инициатора полимеризация не происходит.
Приготовленную реакционную смесь дегазировали в ампулах на вакуумной установке (10–3 мм рт. ст.), после чего ампулы отпаивали и помещали в термостат. В случае распада инициатора при низких температурах (20 °С, УФ) реакционный раствор переносили в кварцевые кюветы (в вакууме).
Сополимеризацию проводили до различных степеней конверсии (исследование полимеризации и сополимеризации до глубоких степеней конверсии может дать важные в практическом отношении результаты), и были выявлены следующие закономерности. Во всех случаях наблюдается образование сополимеров, обогащенных звеньями АГ и МАГ по сравнению с исходной смесью сомономеров (табл. 11), что указывает на большую реакционную способность МАГ в реакциях роста цепи.
Таблица 11
Зависимость состава сополимера от исходного состава реакционного раствора при сополимеризации АГ (МАГ) (М1 ) и ММГ (М2 ) M1 + M2 ] = 2,00 моль/л; [ПСА]= 5·10–3 моль·л–1; Н2 О; 60 °С.
| № п/п | Исходные сомономеры М1: М2, мол.% | Сополимерыа М1: М2, (мол. %)/ [h]б, дл/г | |||
| АГ-ММГ | МАГ-ММГ | ||||
| 1 | 40:60 | 90:10 | 0,35 | 75:25 | 0,15 |
| 2 | 50:50 | 95:5 | 0,55 | 68:32 | 0,20 |
| 3 | 70:30 | 75:25 | 0,88 | 90:10 | 0,27 |
| 4 | 80:20 | 97:3 | 0,93 | 96:4 | 0,41 |
| 5 | 90:10 | 98:2 | 0,98 | 98:2 | 0,53 |
Примечание. а) Определялось по данным ЯМР1 H и ИК-спектроскопии.
б) Определялась при 30 °С в 1н водном растворе NaCl.
На основании исследований радикальной сополимеризации МАГ и ММГ можно сделать вывод, что сополимеризация происходит только при избытке метакрилата гуанидина. Если в избытке находится мономалеинат гуанидина, то ни сополимеризация, ни гомополимеризация метакрилата гуанидина не наблюдается.
Состав синтезированных полимерных продуктов подтвержден методами ЯМР1 Н и ИК-спетроскопии.
Преобладающий вклад стерического фактора в реакционную способность мономалеината гуанидина в реакции сополимеризации с АГ и МАГ подтверждается значениями констант сополимеризации, которые представлены в табл..
Таблица 12
Значение эффективных констант сополимеризации в системах
АГ(МАГ) (М1 ) – ММГ (М2 )
([М]сум = 2 моль×л–1; [ПСА] = 5×10–3 моль×л–1; 60 °С, Н2 О)
| № пп | Сополимеризационная система | r1 | r2 | r1´r2 |
| 1 | АГ-ММГ | 7,82 ± 0.04 | 0,08 ± 0,03 | 0,625 |
| 2 | МАГ-ММГ | 8,97 ± 0.05 | 0,10 ± 0,07 | 0,897 |
3.3 Физико-химические свойства синтезированных сополимеров
Исследования методом ЯМР1 Н и ИК-спектроскопии синтезированных в представленной работе полимерных соединений подтвердили предполагавшуюся структуру объектов исследования. Изучение спектров ЯМР 1 Н синтезированных сополимеров позволило определить сомономерный состав анализом интегральных интенсивностей различных сигналов.
3.3.1 ИК-спектральные исследования синтезированных сополимеров
Анализ ИК-спектральных характеристик проводился сравнением спектров мономерной гуанидисодержащей соли и акриламида, взятыми в качестве моделей, а также сравнением спектров полимерных соединений, которые должны были подтвердить соответствующие изменения в спектрах при переходе от мономеров к сополимерам. ИК спектры всех соединений регистрировали в твердом виде в таблетках KBr.
ИК-спектральные характеристики исходных гуанидинсодержащих мономеров приведены в табл. 13.
Таблица 13
ИК спектральные данные акриловых производных гуанидинаа
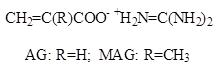
Мономер | Гуанидиновый фрагмент | |||
ν (NH) валентные | ν (C=N) валентные | ν (NH2) деформац. | ν (CNH) углов. дефор. | |
| МАГ | 3100, 3385 | 1680 | 1656 | 520, 544 |
| АГ | З091, 3418 | 1674 | 1660 | 529, 544 |
Мономер | Винильный фрагмент | |||
ν (CH) валентные | ν (C=O) валентные | ν (RC=) скелет. деф. | ν (CH2=C-) неплоск. деф. | |
| МАГ | 2928, 2960 | 1528 | 1240, 1384, 1408, 1456 | 938, 1008 |
| АГ | 2929, 2960 | 1524 | 1275, 1359, 1419 | 956, 988 |
а Положение пиков соответствующих сигналов приведено в см–1 .
При исследовании ИК-спектров сополимеров АГ и МАГ и АА найдено, что в образовавшихся сополимерах присутствуют полосы поглощения характерные для деформационных колебаний связи N-H в акриламиде 1665 см–1 и интенсивные полосы скелетных деформационных колебаний в узле СН3 -С= метакрилатгуанидина при 1470 и 1380 см–1. Причем, в зависимости от состава сополимера интенсивность этих полос меняется. В силу близости строения АА и АГ характеристические полосы сомономеров накладываются и ИК спектры для данной пары недостаточно информатины. В спектрах присутствует также полоса поглощения карбоксилат-иона (1560-1520 см–1 ). Полосы валентных колебаний N-H связей сильно сдвинуты в сторону длинных волн (3130 и 3430 см–1 ) и являются достаточно интенсивными. В спектре сополимера присутствует интенсивная широкая полоса с максимумом при 1648 см–1, которая, конечно, искажена поглощением деформационных колебаний воды в этой области, но интенсивность ее и наличие нескольких перегибов на плечах свидетельствует о том, что в данном соединении присутствует и связь N=C и NH2 группа.
Характерные для углеводородных цепей с полярными концевыми группами крутильные колебания СН2 -групп проявляются в области 1180- 1320 см–1 .
Для определения содержания СН3 — групп использовали полосу поглощения 1380 см-1, относящуюся к симметричным деформационным колебаниям. Другие полосы, характеризующие метакрилатный анион, также хорошо проявляются в спектре: 2960, 2928 см-1 (валентные колебания CH связей) (рис. 10-13).
![]()
Рис. 10. ИК-спектр полиметакрилатгуанидина
Рис. 11. ИК-спектр сополимера АА-МАГ (50:50)
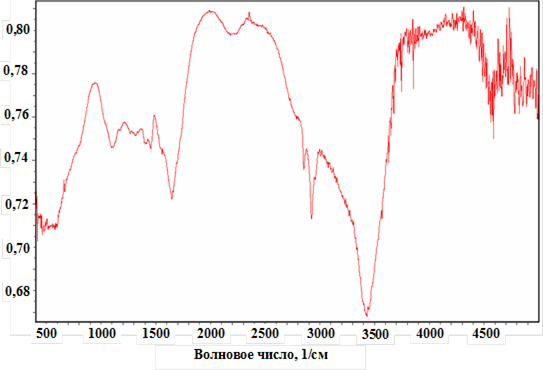
Рис. 12. ИК-спектр сополимера АА-МАГ(90:10)
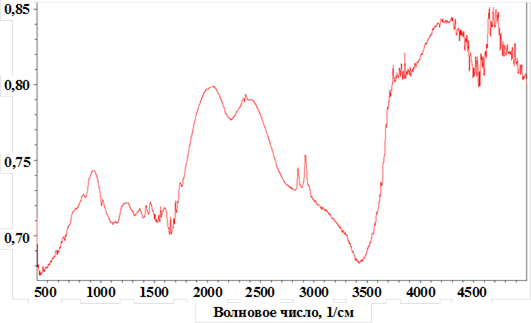
Рис. 13. ИК-спектр сополимера АА-МАГ (30:70)
ИК-спектры сополимеров ММГ с МАГ характеризуются наличием полосы поглощения 1170 см–1 характерной для малеинатов и полосы 1630 см–1 монозамещенного гуанидиния. Две интенсивные полосы 1680 см–1 и 1656 см–1 связаны с C=N валентными колебаниями и смешанными с ними деформациями NH2 групп. Колебания карбонильной группы монозамещенной малеиновой кислоты появляются на спектре в области 1730 см–1, ярко выражены полосы поглощения метильных групп (1380-1460 см–1 ) интенсивность которых также меняется в зависимости от состава сополимера.
3.3.2 ЯМР-спектральные характеристики сополимеров акриламида и метакрилата гуанидина
В данном разделе приводятся ЯМР-спектральные характеристики синтезированных сополимеров. При изучении спектров протонного магнитного резонанса в качестве модельных соединений использовали метакриловую кислоту, акрилат и метакрилат гуанидина, акриламид.
Спектры ЯМР 1 Н акриловой кислоты (АК) и ее гуанидиновой соли АГ относятся к АВС типу, характеристики сигналов суммированы в табл.14.
Отметим небольшое смещение в более сильное поле сигналов метиленовых протонов (3 С) АГ в сравнении с АК. По всей видимости, это связано с тем, что для АГ в воде (схема 13) более характерна структура односвязанного водородного комплекса и (или) димера, что лишь в незначительной степени снижает дезэкранирующее действие карбоксильной группы. С другой стороны, сигналы протона у 2 С в спектре АГ смещены в слабое поле по сравнению с АК; вероятно, это может быть связано с изменением в растворе конформации АГ в сравнении с АК, и протон у 2 С переместится из положительной области конуса анизотропии С=О группы в отрицательную область.
Таблица 14
Спектральные характеристики акрилатных производных а, б .
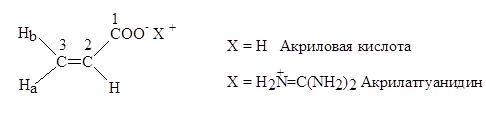
| Соединение | Растворитель | 3Ha | 3Hb | 2H | NH | ||||||
| δ 3a | n | J 3a,. 2 | J 3a,. 3b | δ 3б | n | J 3a, 2 | δ 2 | n | δ | ||
| АК | D2О | 5,94 | 4 | 9,63 | 1,05 | 6,36 | 4 | 17,13 | 6,11 | 4 | – |
| АГ | D2О | 5,91 | 4 | 9,64 | 2,14 | 6,27 | 4 | 17,67 | 6,41 | 4 | – |
| АГ | ДМСО | 5,28 | 4 | 9,64 | 3,21 | 5,79 | 4 | 17,14 | 5,96 | 4 | 7,74 |
Примечания: а Основные сокращения: δ – величина химического сдвига соответствующих протонов, в м.д.; n – число линий в сигнале данного типа протонов; Jij – константы спин-спинового взаимодействия соответствующих протонов, в Гц. б Число протонов по интегральным интенсивностям согласуется с предполагаемой структурой: по 1Н для всех протонов винильной системы и 6Н для гуанидинового противоиона (проявляется уширенным синглетом).
Схема 8
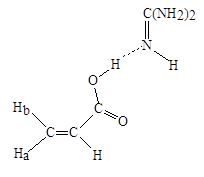 |  |
Спектры ЯМР 1 Н метакриловой кислоты и ее гуанидиновой соли МАГ относятся к АВХ3 типу, характеристики сигналов суммированы в табл. 15; во всех случаях не наблюдалось полного расщепления сигналов, т.е. имелся вырожденный АВХ3 тип спектров.
Таблица 15
Спектральные характеристики метакрилатных производных а, б .
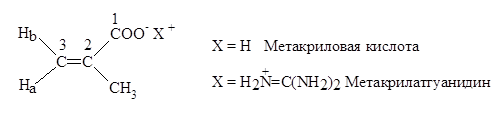
| Соединение | Растворитель | CH3 | 3Ha | 3Hb | NH | ||||||
| δ | n | J Me,3a | J Me, 3b | δ 3a | n | J 3a,Me | δ 3b | n | δ | ||
| МАК | D2О | 2,10 | 3 | 1,62 | 1,05 | 5,90 | 3 | 1,62 | 6,31 | 1 | – |
| МАГ | D2О | 2,08 | 3 | 2,13 | 1,08 | 5,53 | 1 | – | 5,86 | 1 | – |
| МАГ | ДМСО | 1,77 | 1 | – | – | 5,04 | 3 | 1,62 | 5,60 | 2 | 7,67 |
Примечания: а Основные сокращения: δ – величина химического сдвига соответствующих протонов, в м.д.; n – число линий в сигнале данного типа протонов; Jij – константы спин-спинового взаимодействия соответствующих протонов, в Гц. б Число протонов по интегральным интенсивностям согласуется с предполагаемой структурой: по 1 Н – для метиленовых протонов, 3Н – для метильных протонов и 6Н для гуанидинового противоиона (проявляется уширенным синглетом).
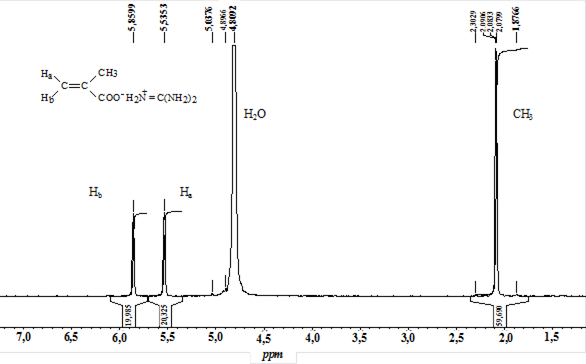
Рисунок 14. ЯМР1 Н спектр метакрилатгуанидина в D2 O
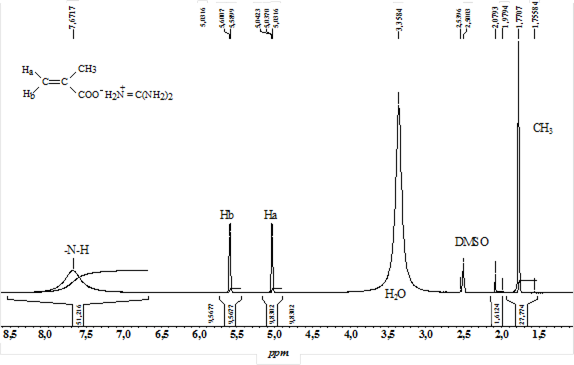
Рисунок 15. ЯМР1 Н спектр метакрилатгуанидина в ДМСО-d6
Отметим, что во всех случаях не наблюдалось полного расщепления сигналов, т.е. имелся вырожденный АВХ3 тип спектров. Это может быть связано с сильным влиянием СООХ группы (особенно в случае МАГ).
Спектры ЯМР1 Н новых сополимеров АГ и МАГ с ААм характеризуются уширенными, неразрешенными (обычными для полимерных структур) сигналами СН2 — и СН-групп цепи и боковых СН3 – групп в случае МАГ. В случае АГ в связи с близостью химических сдвигов протонов СН2 -СН= в обоих сомономерах, то разделить их вклад по сомономерам не удается (рис. 16,17).
Рисунок 16. ЯМР1 Н спектр сополимера АГ-ААм (80:20) в D2 O
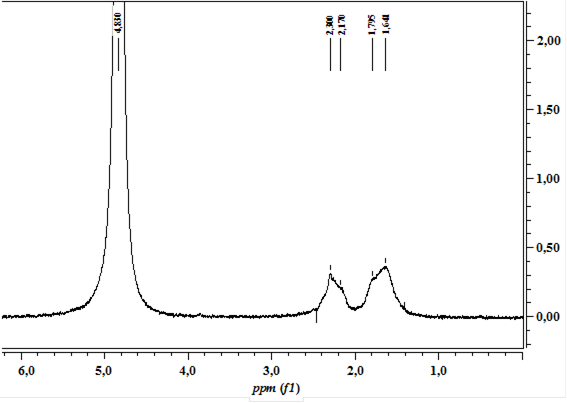
Рисунок 17. ЯМР1 Н спектр сополимера АГ-ААм (40:60) в D2 O
В сополимерах, обогащенных акриламидным сомономером, сигналы звеньев МАГ смещаются в более слабое поле. В сополимерах, обогащенных сомономером МАГ, сигналы звеньев АА смещаются в более сильное поле. Это можно объяснить образованием внутри- и межмолекулярных водородных связей между боковыми группами амидной и гуанидиновым противоионом. Это усиливает дезэкранирование для звеньев МАГи экранирование для звеньев АА.
Таблица 16
Спектральные характеристики сополимеров АА(М1 ) – МАГ (М2 ) и соответствующих гомополимеров (ПААм и ПМАГ), измеренные в D2 O (в м. д.).
| Соединение | Исходный состав m1: m2, мол% | M1 | M2 | ||
| CH2 | CH | CH3 | CH2 | ||
| ПАА | 100: 0 | 1,58; 1,73; 1,85 | 2,27; 2,42 | – | – |
| ПМАГ | 0: 100 | – | – | 1,01; 1,05 | 1,74 |
| СП | 90: 10 | 1,57; 1,73; 1,85 | 2,28; 2,42 | 1,11 | закрыт |
| СП | 80: 20 | 1,57; 1,73; 1,85 | 2,27; 2,40 | 1,09 | закрыт |
| СП | 50: 50 | 1,69 | 2,19 | 1,06 | 1,69 |
| СП | 30: 70 | 1,49; 1,65 | 2,22 | 1,02 | 1,80 |
| СП | 10: 90 | – | – | 1,06 | 1,76 |
Расчет состава сополимеров проводили, используя интегральную интенсивность сигнала метильной группы сомономера МАГ (рис. 18, 19), который проявляется в самом сильном поле и не перекрывается никакими другими сигналами по методике, указанной выше.
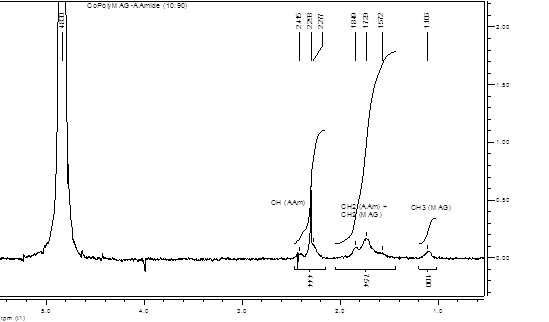
Рис. 18. ЯМР1 Н спектр сополимера МАГ-АА (10:90) в D2 O

Рис. 19. ЯМР1 Н спектр сополимера МАГ-АА (70:30) в D2 O
ЯМР1 H-спектры сополимеров АГ и МАГ с мономалеинатом гуанидина (рис. 20, 21) свидетельствуют об обогащении сополимеров АГ и МАГ.

Рис. 20. ЯМР1 Н спектр сополимера АГ-ММГ (70:30) в D2 O

Рис. 21. ЯМР1 Н спектр сополимера МАГ-ММГ (70:30) в D2 O
3.3.3 Термические свойства синтезированных сополимеров
Стойкость соединений, в том числе полимерных, к воздействию различных температур является важной характеристикой веществ, которые предполагается использовать в составе различных композиций.
Для изучения термофизических свойств синтезированных продуктов и исходных реагентов использовали программно-аппаратный комплекс с пакетом компьютерных программ, предназначенных для количественной обработки дериватограмм (кривых Г, TG, DTG, DTA), разработанный в Институте химии растворов РАН (г. Иваново) для измерения и регистрации выходных сигналов от датчиков дериватографа 1000D (MOM, Венгрия).
На рис. 22 представлены ТГ-кривые сополимера АА с МАГ 50:50 на воздухе. Потеря веса сополимером наблюдаются при температуре 150 °С; по-видимому, это связано с потерей воды и удалением летучих примесей. Уменьшение массы на 10% наблюдается при температуре 150 ºС. Скорость термического и термоокислительного разложения сополимера заметно возрастает при температуре 210 °С. Выше этой температуры можно отметить две стадии разложения: 250-300 °С и 300-390 °С; эндотермический эффект при температуре 390 °С, который при 520 ºС переходит в экзоэфект, отражающий термоокислительную деструкцию полимера. Выше 600 ºС происходит удаление коксовой массы и остается 8% твердого остатка. Общее падение массы составляет 80 %.
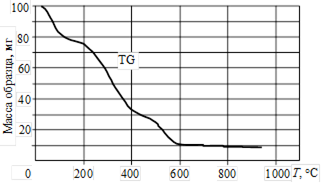
Рис.22. Зависимость потери массы от температуры сополимера АА-МАГ (50:50)
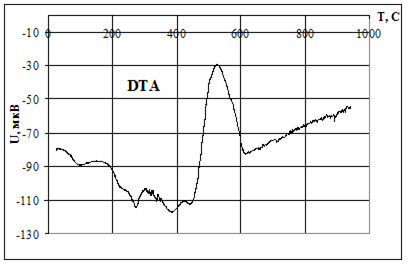
а)
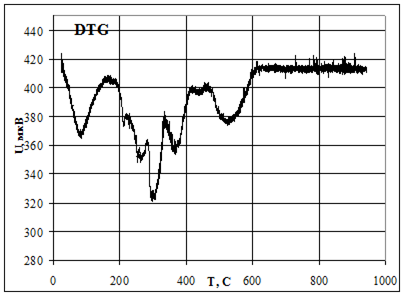
б)
Рис. 23. Кривые ДТА(а) и ДТГ (б) сополимера АА-МАГ (50:50)
Рассмотрим термостабильность сополимера с большим содержанием метакрилата гуанидина МАГ-АА (90:10)
Как видно на кривой ТГ, потеря массы, связанная с удалением воды и летучих примесей из образца, наблюдается в области температур от 150 до 240 ºС, при этом потеря массы составляет до 15 %. Далее идет стремительное уменьшение массы до температуры 570 ºС. На этом участке происходит разложение гуанидиновых остатков, в результате дальнейшее разложение идет с образованием летучих продуктов, что приводит к вспениванию исследуемых образцов. При этой температуре на кривой ДТА наблюдается экзотермический эффект, показывающий полное термоокисление полимера. После удаления коксовой массы остается 20 % твердого остатка.
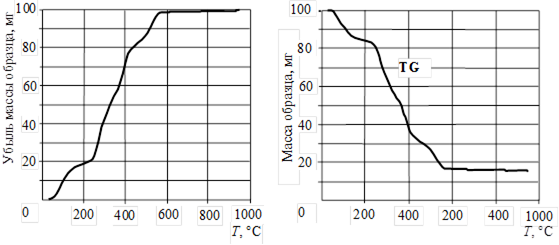
Рис. 24. Зависимость потери массы от температуры сополимера АА-МАГ (90:10)
При анализе кривых ТГ выявлено, что масса твердого остатка выше в образцах с большим содержанием МАГ.
По данным ДСК оказалось, что в образцах гомо- и сополимеров, взятых для исследований, воды около 20%, т.е. такая характеристика термостабильности соединений, как потеря 10% массы, требует корректировки данных ДТА для полимерных соединений. При этом следует отметить, что вода в сополимерах связана прочнее, чем в ПМАГ: при исследовании методом ДСК прогрев образцов ПМАГ до температуры 150 °С с последующим охлаждением и новым нагревом показал, что вода из данного соединения удалена полностью, чего не удалось добиться для сополимеров.
Наиболее стабильными оказались образцы сополимеров, содержащих большее количество акриламида. Например, потеря 30 % массы для сополимера АА-МАГ (90:10) наблюдается при 300 °С, а для сополимера 30:70 – при 280 °С. Это, вероятно связано с более сложным строением сополимеров с большим содержанием метакрилата гуанидина. По данным работы [171] при термоокислении производных мочевины, в том числе и гуанидина, могут выделяться водород, угарный газ, углекислый газ, метан.
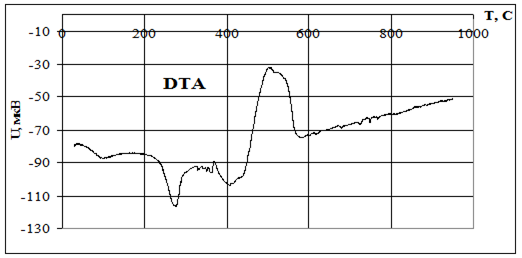
а)
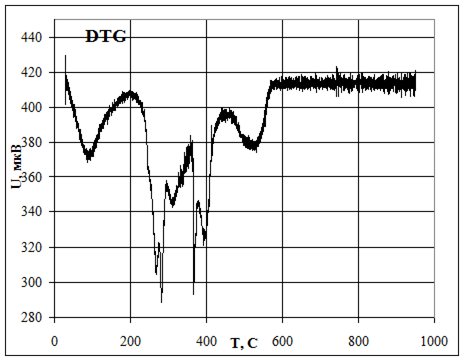
б)
Рис. 25. Кривые ДТА(а) и ДТГ (б) сополимера АА-МАГ (10:90)
С учетом возможного термолиза гуанидина с образованием карбамида суммарная реакция термодеструкции гуанидинового остатка упрощенно может быть представлена следующей реакцией:
72СО(NH2 )2 → 45NH3 + 15CO + 15H2 O + 5N2 + 4CO2 + 17(NH2 )2 (CO)2 NH +19NH2 CN
Сополимеры акриламида оказались более термостабильными, чем полиакриламид. Полиакриламид термически устойчив до 130 °С, а потеря 30% массы наблюдается уже при температуре 170 °С. При более высоких температурах начинается деструкция полимера, которая, как известно [172], сопровождается выделением аммиака, образованием имидных групп, возникновением внутри- и межмолекулярных связей по типу:
Таким образом, при сравнении термостабильности полимерных продуктов можно отметить, что более стабильными во всем интервале температур оказались сополимеры в сравнении с гомополимерами.
Данные термофизических исследований синтезированных сополимеров АГ и МАГ с ММГ суммированы в табл. 17 и 18.
Таблица 17
Термофизические свойства исходных мономеров и сополимеров МАГ-ММГ
Сополимеры МАГ: ММГ | кривая ДТА, T пл | кривая. ДТГ интервал разложен. | Ум-е массы на 10% | Ум-е массы на 20% | Ум-е массы на 30% | Мост. |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| 90:10 | 200 350 500 540 | 80 280 400 520 | 110 | 270 | 300 | 5% |
| 60:40 | 370 500 550 | 80 260 280 350 410 515 | 100 | 250 | 290 | 7% |
| 50:50 | 310 370 490 540 | 80 270 340 400 520 | 100 | 250 | 290 | 8% |
| 10:90 | 150 550 640 | 80 550 | 200 | 410 | 470 | 12% |
| МАГ | 167 168 | 202-242 270-357 | 222 | 320 | 370 | 9% |
| ММГ | 110 150 175 240 290-390 610 740 | 75-155 155-170 170-240 240-550 | 150 | 180 | 230 | 6% |
Таблица 18
Термофизические свойства исходных мономеров и сополимеров АГ – ММГ
| Образец | кривая ДТА Tпл | кривая ДТГ интервал разложен. | Уменьшение массы на 10% | Уменьшение массы на 20% | Уменьшение массы на 30% | Мост. |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| АГ | 90 270 370 540 640 | 90-160 160-205 205-260 260-350 350-450 450-570 | 70 | 100 | 230 | 6% |
| ММГ | 110 150 175 240 290-390 610 740 | 75-155 155-170 170-240 240-550 | 150 | 180 | 230 | 8% |
АГ–ММГ 50:50 | 170 270 350 600 | 90-185 185-265 265-330 330-520 | 150 | 110 | 250 | 12% |
АГ–ММГ 70:30 | 270 360 560 600 | 170-200 200-250 250-440 440-560 | 100 | 200 | 250 | 10% |
Таким образом, исследование термостабильности сополимеров показало, что их термические свойства зависят от состава и значительно выше термических характеристик исходных сомономеров и гомополимеров.
3.4. Исследование бактерицидных и токсикологических свойств новых сополимеров акрилат- и метакрилатгуанидина
В настоящий момент трудно найти группу материалов, на которую микроорганизмы не оказывают разрушающего действия. Жизнедеятельность различных патогенных микробов вызывает не только нежелательные изменения структурных и функциональных характеристик материалов и изделий, но они также реализуют свое губительное действие внутри живых клеток организма. В связи с этим, разработка новых биоцидных препаратов, несомненно, является актуальной задачей.
Учитывая, что под собственной физилогической активностью полимеров обычно понимают активность, которая связана с полимерным состоянием и не свойственна низкомолекулярным аналогам или мономерам [174], механизмы проявления собственной физиологической активности могут включать в себя как важнейшую составляющую физические эффекты, связанные с большой массой, осмотическим давлением, конформационными перестройками и др., а также могут быть связаны с межмолекулярными взаимодействиями и с биополимерами организма. Многие биополимеры организма являются полианионами (белки, нуклеиновые кислоты, ряд полисахаридов), а биомембраны также имеют суммарный отрицательный заряд. Взаимодействие между противоположно заряженными полиэлектролитами протекают кооперативно, причем образующиеся в результате поликомплексы достаточно прочны. Известно, что наибольшее значение имеют при таких взаимодействиях плотность заряда и молекулярная масса [174]. Если же говорить о биоцидных свойствах, то важную роль в этом случае играет знание механизма действия.
Последовательность элементарных актов летального действия полиэлектролитов на бактериальные клетки может быть представлена следующим образом [194]:
1) адсорбция поликатиона на поверхности бактериальной клетки;
2) диффузия через клеточную стенку;
3) связывание с цитоплазматической мембраной;
4) разрушение или дестабилизация цитоплазматической мембраны;
5) выделение из клетки компонентов цитоплазмы;
6) гибель клетки.
В первую очередь, это касается поликатионов, так как биомембраны имеют отрицательный суммарный заряд, хотя, отрицательно заряженные в целом клеточные мембраны имеют изолированные поликатионные области, на которых могут сорбироваться полианионы [195].
Все вышесказанное свидетельствует о перспективности и принципиальной возможность использования в качестве биоцидных препаратов синтезированных нами гуанидинсодержащих полимерных веществ. Отметим, что эти полимеры отвечают ряду требований, которые предъявляются к современным препаратам подобного рода: хорошая растворимость в воде и физиологическом растворе (1%-е растворы полимеров имеют рН =6,5-7,0); растворы бесцветны, не имеют запаха, не вызывают разрушения обрабатываемых материалов, а также полимерная природа этих соединений способствует отсутствию ингаляционной токсичности и образованию на обработанных поверхностях длительно сохраняющейся полимерной пленки, обеспечивающей пролонгированный биоцидный эффект.
Как известно, радикальная сополимеризация акриламида с виниловыми мономерами используется для получения сополимеров, которые обладают лучшими потребительскими свойствами по сравнению с полиакриламидом, который является промышленным флоккулянтом и используется в самых разных отраслях промышленности.
Предполагалось, что сополимеры АА, содержащие гуанидиновые группы будут обладать не только флоккулирующими, но биоцидными свойствами.
Биоцидную активность определяли по методикам подсчета выросших колоний после обработки воды флоккулянтами и методом диффузии в чашке (см. экспериментальную часть).
В результате исследований было выявлено, что полученные сополимеры обладают значительной биоцидной активностью по отношению к кишечной палочке, при этом биоцидная активность повышается с увеличением содержания гуанидинового фрагмента.
Таблица 19
| Полимеры и сополимеры* | Число колоний, выросших на 1 см3 воды | Качественная оценка биоцидности |
| 1 | 1206 | неудовлетворительная |
| 2 | 31 | хорошая |
| 3 | 35 | хорошая |
| 4 | 103 | удовлетворительная |
*Примечание. 1-полиакриламид, 2-сополимер АА: МАГ (70:30),
3-сополимер АА: АГ (80:20), сополимер АА: МАГ (90:10).
Таблица 20
| Полимеры и сополимеры* | Диаметр зоны задержки роста (мм) | Качественная оценка биоцидности |
| 1 | 2 | неудовлетворительная |
| 2 | 15 | отлично |
| 3 | 10 | хорошая |
| 4 | 6 | удовлетворительная |
Как видно из полученных результатов, синтезированные гуанидинсодержащие сополимеры проявляют бактерицидную активность в отношении изученных клеточных структур, причем у сополимеров с большим содержанием гуанидиновых групп наблюдается наиболее выраженная биоцидная активность.
На бактериологической стации ГСЭН КБР исследована также биоцидная активность сополимеров относительно золотистого стафилококка и патогенной грибковой микрофлоры Candidaalbicans.
Выявлено, что наибольшей биоцидной активностью по отношению к золотистому стафилококку обладает сополимеры АА-МАГ (70:30), (50:50), (10:90). Видно, что биоцидная активность зависит от количества МАГ в макромолекулярной цепи. По отношению к Candidaalbicans наиболее активным оказались образцы АА-МАГ (10:90) и АА-АГ (20:80). АА-МАГ (10:90).
Одним из важных показателей для применения реагента в качестве флоккулянта являются его токсикологические характеристики, так как для очистки воды могут применяться полимеры, не действующие на человека, животных, фауну и флору водоемов.
Методы биотестирования на ветвистоусых ракообразных занимают ведущее положение в системе экологического мониторинга природных вод, а биотест на дафниях Daphnia magma Strauss является наиболее стандартизованным из всех известных [196-198]. При биотестировании природных вод на зоопланктоне регистрируют поведенческие реакции, патологические нарушения, метаболические (биохимические) показатели, физиологические функции, окраску тела, скорость выедания корма и др., но наиболее чувствительной и надежной считается тест-реакция, в которой регистрируется процессы размножения – выживаемость и плодовитость.
Для определения токсичности ряда гомо- и сополимеров использовали методику определения токсичности воды с помощью дафний Daphnia magma Strauss. Дафний в количестве 20 штук высаживали в чашки Петри с исследуемыми образцами. Контроль проводили визуально и с применением бинокуляра, контролируя количество выживших дафний, причем учитывались изменения в движении и размножении рачков. Параллельно ставили контрольный опыт с природной водой. Наблюдения проводили 96 часов, дафний во время эксперимента не кормили. По окончании эксперимента проводили учет выживших дафний, выжившими считаются дафнии, если они свободно передвигаются или всплывают со дна.
Коэффициент токсичности в % рассчитывали по формуле:
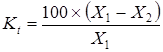 , (10)
, (10)
где, Х1 и Х2 среднее арифметическое количество выживших дафний в контроле и опыте.
Проба воды оценивалась как обладающая острой токсичностью, если за 96 часов биотестирования в ней гибло 50% и более дафний по сравнению с контролем.
Токсикологические характеристики сополимеров исследовали в зависимости от состава и концентрации при постоянной температуре. В качестве модельных образцов были взяты соответствующие гомополимеры – полиакриламид и полиметакрилат гуанидина.
Растворы гомополимеров и сополимеров без разбавления оказывают угнетающее действие на весь процесс размножения дафний (рис. 26), задерживает рост, наступление половой зрелости и появление первого помета, уменьшает количество пометов и плодовитость, повышает выброс молоди и яиц. При разведении в отношении 1:2 токсичность сополимеров снижается. Наименее токсичными являются растворы сополимеров с концентрацией от 0,1 до 0,01 %. Токсичность образцов зависит также от состава сополимеров; с увеличением содержания метакрилата гуанидина токсичность снижается.
Анализ экспериментальных данных по исследованию токсичности сополимеров показывает, что растворы сополимеров МАГ: АА (20:80) и МАГ: АА (30:70) с концентрацией 0,1% и 0,01% практически не влияют на плодовитость дафний, но на 15% сокращают длительность жизни. Отметим, что гомополимер ПМАГ снижает плодовитость и длительность жизни у исследуемых дафний всего на 7%, а полиакриламид на 30 %. Выявлено, что токсичность полиакриламида выше, чем у сополимеров, т.е. даже небольшое содержание метакрилата гуанидина в сополимерах уже снижает токсичность полиакриламидного флоккулянта.
Рис. 26. Зависимость коэффициента токсичности гомо- и сополимеров от состава и концентрации.
Как известно, результаты биотестирования зависят от чувствительности тест-организмов. Поэтому помимо D. magna, для токсикологической оценки водных растворов полимерных флоккулянтов использовали также личинок комаров — звонцов Chironomusdorsalis. Результаты анализа показали, что наименее токсичными в исследованных условиях являются сополимеры АА с МАГ по сравнению с ПАА, причем наименее токсичным образцом для данных тест-культур оказался сополимер АА: МАГ (70:30), в растворе которого наблюдался переход личинок в куколки, а затем превращение в имаго. Исследование токсичности АА с АГ показало, что данные сополимеры обладают еще меньшей токсичностью по сравнению с МАГ, что хорошо согласуется с литературными данными о меньшей токсичности акриловой кислоты по сравнению с метакриловой.
Учитывая полученные данные можно варьировать состав сополимеров для достижения максимального эффекта биоцидного действия при минимальных проявлениях токсичности. Наличие же в структуре синтезированных сополимеров химически активных гуанидиновых групп открывает возможность осуществления на их основе макромолекулярного дизайна, что расширит области практического применения исследуемых сополимеров.
Таблица 21
Данные по биоцидности и токсичности сополимеров АГ и МАГ с ММГ и ряда модельных полимеров а
| № пп | Соединение | М1: М2 (исходный состав) | It | E. coli | St.aureus | Candida albicans | МПК ´103 д |
| 1 | ПМАК | - | 93,2 | --- | --- | --- | - |
| 2 | ПАГ | - | 76,1 | --- | -++ | --- | - |
| 3 | ПМАГ | 0:100 | 104,6 | --- | -++ | --- | 9,2 |
| 4 | АГ-ММГ | 50:50 | 88,7 | --- | --- | +++ | 2,2 |
| 5 | МАГ-ММГ | 70:30 | 101,5 | --- | ---- | ++++ | 2,7 |
| 6 | МАГ-ММГ | 50:50 | 119,1 | --- | ---- | ++++ | 1,4 |
![]() Примечания. Escherichia coli – кишечная палочка, представитель грамотрицательной бактерии и Stophil. Aureus 906 – золотистый стафилококк, представитель грамположительной бактерии; (+++) – сплошной лизис бактериальной клетки, полностью задерживает рост данного штамма, (--+) — – частичный лизис клетки, наблюдаются зоны подавления роста через 48 часа (--+) – частичный лизис клетки, наблюдаются зоны подавления роста через 72 часа, (---) – не активен. д Минимальная подавляющая концентрация в вес%.
Примечания. Escherichia coli – кишечная палочка, представитель грамотрицательной бактерии и Stophil. Aureus 906 – золотистый стафилококк, представитель грамположительной бактерии; (+++) – сплошной лизис бактериальной клетки, полностью задерживает рост данного штамма, (--+) — – частичный лизис клетки, наблюдаются зоны подавления роста через 48 часа (--+) – частичный лизис клетки, наблюдаются зоны подавления роста через 72 часа, (---) – не активен. д Минимальная подавляющая концентрация в вес%.
Сополимеры АГ и МАГ с ММГ не активны по отношению к изученным микроорганизмам, но обладают высокой фунгицидной активностью по отношению к патогенной грибковой микрофлоре Candidaalbicans, примечательно, что соответствующие гомополимеры проявляют бактерицидную активность, а фунгицидной активностью не обладают. Так, наибольший противогрибковый эффект был получен для образцов сополимеров МАГ с ММГ с исходным составом сомономеров 50:50 и 70:30.
Таким образом, сочетание в полученных сополимерах высокой противогрибковой активности (за счет содержания гуанидиновых групп) с повышенной способностью связываться с бактериальными клетками, благодаря звеньям гуанидина, позволило нам синтезировать новые эффективные гуанидинсодержащие биоцидные полимеры.
3.5 Исследование флоккулирующих свойств новых сополимеров акриламида
Одним из наиболее широко применяемых методов снижения количества взвеси является седиментация под воздействием сил тяжести частиц. Поскольку частицы взвеси, обусловливающие мутность природных вод, отличаются малыми размерами, их осаждение происходит крайне медленно; кроме того, наличие примесей коллоидного характера еще более осложняет процесс седиментации.
Для интенсификации процесса осаждения и повышения его эффективности применяется обработка воды коагулянтами. Несмотря на большую эффективность, технология очистки воды, основанная на применении коагулянтов, обладает рядом недостатков. Важнейший из них – малая прочность хлопьев, образующихся при коагуляции, не позволяющая работать при высоких скоростях потока воды и приводящая к выносу загрязнений из фильтующей загрузки [173]. При применении высокомолекулярных флоккулянтов устраняются основные недостатки коагулирования, повышается прочность хлопьев и ускоряется процесс их образования. Это позволяет увеличить эффективность осветления воды: сократить время отстаивания, повысить производительность осветлителей с взвешенным осадком, увеличить грязеемкость фильтров и контактных осветлителей.
В настоящее время сополимеры акриламида являются наиболее распространенными флоккулянтами. В связи с этим синтез и исследование флоккулирующих свойств новых сополимеров акриламида является, несомненно, актуальной задачей.
Обычно определение эффективности флоккулянтов по отношению к определенному виду загрязняющих воду веществ заключается в определении концентрации этих веществ в воде до и после обработки флоккулянтами.
Для оценки флоккулирующей активности полиэлектролитов необходимо использование модельных систем. В качестве моделей чаще всего используют водные суспензии каолина, охры и бентонита. Причем именно на суспензиях каолина описаны закономерности флоккулирующего действия большого числа катионных полиэлектролитов [174–177, 179–181]. В литературе также отмечается [182], что при концентрации каолина ~ 0,8 % и ниже частицы суспензии способны осаждаться в свободном режиме, и в этих условиях результаты экспериментов могут использоваться для изучения закономерностей флоккуляции.
Так как на флоккулирующую способность оказывает влияние величина заряда макромолекулы, то для исследования выбрали сополимеры с различной степенью содержания звеньев метакрилата гуанидина в макромолекулярной цепи. В качестве объекта сравнения использован полиакриламид. Флоккулирующую активность исследовали как в присутствии и отсутствии коагулянтов. В качестве коагулянта использовали органомодифицированную глину месторождения Герпегеж.
На рис. 27. показано влияние концентрации флоккулянтов разного состава на флоккулирующий эффект (F), который рассчитывали по формуле (11)
F = (n0 — n) / n, (11)
где n0и n — соответственно оптическая плотность воды (определена турбидиметрическим методом) в отсутствие и в присутствии флоккулянта (и коагулянта).
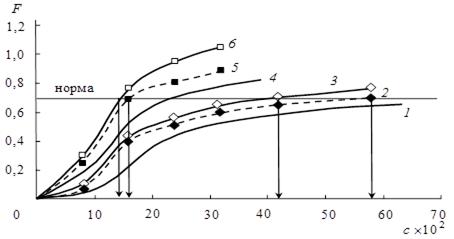
Рис.27. Зависимость флоккулирующего эффекта F от концентрации и состава сополимеров 1- ПАА; 2- АГ-АА (20:80); 3- АГ-АА (40:60); 4- МАГ-АА (20:80); 5- МАГ-АА (40:60); 6- МАГ-АА (30:70)
Опыты, проведенные на одной партии природной воды (мутность 4,2 мг·л–1, цветность 48,5 градусов) показали увеличение флоккулирующего эффекта с ростом концентрации сополимера для всех флоккулянтов. Это следствие увеличения концентрации макромолекулярных мостиков, образованных при адсорбции макромолекул на поверхности частиц дисперсной фазы, что формировало крупные агрегаты из частиц дисперсной фазы и макромолекул и снижало устойчивость системы.
Также на рисунке видно, что образцы сополимера АА: МАГ (кривые 4,5,6) характеризуются большими величинами F по сравнению с ПАА (кривая 1). Сопоставление данных рис. 27. при постоянной концентрации флоккулянтов, свидетельствует о возрастании значений F при переходе к сополимерам с более высоким содержанием звеньев АГ и МАГ (кривые 2-6).
Из рис. 27 также следует, что отвечающий норме D = 0,7 (определен при n = 0,172 и λ = 364 нм, соответствующей мутности очищенной воды) достигается при меньших значениях концентрации сополимера АА: МАГ по сравнению с ПАА.
На рисунке видно, что максимальный флоккулирующий эффект наблюдается у сополимера состава 70:30. Очевидно, при этом реализуется оптимальное соотношение между плотностью заряда и гибкостью макромолекул, которое обеспечивает охват полимерными мостиками большего числа частиц дисперсной фазы, увеличению размера флоккул и D.
Видно, что образцы сополимеров МАГ-АА характеризуются большими величинами F по сравнению с АГ-АА, поэтому более подробно изучены флоккулирующие свойства сополимера МАГ-АА.
Несомненное влияние на процесс флоккуляции должен оказывать размер макромолекул или молекулярная масса полимера. Чем больше размер макромолекул, тем относительно больший процент сегментов адсорбированных макромолекул остается свободным и способным к адсорбции на других частицах. Большая макромолекула может связать большее число твердых частиц, образуя, таким образом, более крупные хлопья.
Для выяснения влияния молекулярной массы на степень флоккулирующего действия нами были исследованы образцы сополимеров с различными молекулярными массами. О величине молекулярной массы судили по характеристической вязкости растворов сополимеров.
Таблица 22
Влияние характеристической вязкости на процесс
осветления модельного раствора
Сополимер АА: МАГ | [η], дл/г | концентрация сополимера,% | Степень осветления |
| 70:30 | 3,2 | 0,05 | 75,8 |
| 70:30 | 2,2 | 0,05 | 95,8 |
| 70:30 | 1,6 | 0,05 | 72,4 |
| 60:40 | 2,8 | 0,05 | 77,5 |
| 60:40 | 1,7 | 0,05 | 82,8 |
| 60:40 | 1,0 | 0,05 | 66,9 |
Рассматривая влияние молекулярной массы полиэлектролитов, было обнаружено, что наибольшие скорости и степени осветления суспензии получены с использованием сополимера, имеющего промежуточное значение молекулярной массы. Образцы с меньшей и с большей молекулярной массой проявляют несколько пониженную активность.
Некоторое снижение скорости осветления и степени осветления с ростом молекулярной массы вероятно связано с влиянием диффузионных ограничений, которые влияют на распределение макромолекул по частицам дисперсии. Особенно эффект снижения эффективности осветления проявляется для сополимера с наиболее высокими значениями характеристической вязкости. Хотя скорость осветления для этих сополимеров выше в очень широком диапазоне концентраций, что указывает на формирование крупных флоккул, степень осветления не превышает 76 %.
Видимо, в системе остается достаточно большое количество несфлоккулированных частиц. Вероятно, по мере возрастания размеров макромолекул усиливаются стерические явления и затрудняется подход частиц с адсорбированными макромолекулами к свободной поверхности других частиц.
Причины невозможности флоккуляции в случае больших размеров макромолекул объяснены в работе [199]. Авторы отмечают, что при большом различии в размерах коллоидных частиц и макромолекул полимера флоккуляция вообще становится невозможной вследствие малой вероятности образования полимерных мостиков, что наглядно показано на рис. 28.
а) б)
Рис. 28. Влияние соотношения размеров макромолекул и коллоидных частиц на процесс флоккуляции: а) макромолекулы намного больше коллоидных частиц; б) коллоидные частицы намного больше макромолекул; h- статистический размер макромолекул, d-размер коллоидных частиц.
Таким образом, для флоккуляции необходимо, чтобы молекулы полимера и твердые частицы приближались друг к другу на расстояние, достаточное для осуществления адсорбции и образования полимерных мостиков.
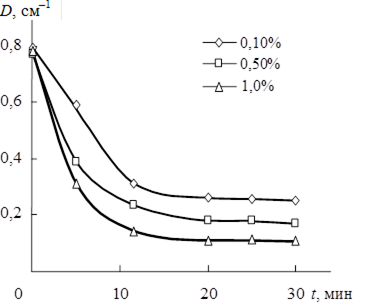
Рис. 29. Зависимость оптической плотности суспензии каолина от времени отстаивания и концентрации сополимера состава 70:30
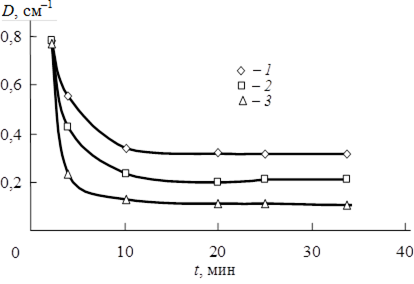
Рис. 30. Зависимость оптической плотности суспензии каолина
от времени отстаивания и состава флоккулянта
Сочетание высокой скорости осветления и наибольшей степени осаждения частиц достигается при использовании сополимера акриламида с метакрилатом гуанидина состава 70:30. Так в интервале доз полиэлектролита 0,05 – 0,12 масс.% максимальная эффективность осаждения составляет 95 – 96%. Оптимальные концентрации полиэлектролитов на основе сополимеров АА: МАГ, исходя из турбидиметрических кривых, составляют 0,5 – 1,0%.
Для изучения механизма образования флоккул и осадков необходимо использование методов, непосредственно характеризующих кинетическую и агрегативную устойчивость флоккулированных дисперсий. К таким методам относятся определение кинетических параметров осаждения дисперсий.
На рис. 31 представлены кинетические кривые осветления суспензии каолина с концентрацией 0,5 масс. %.
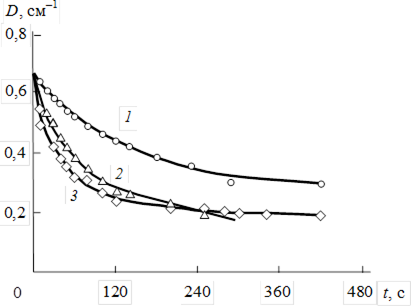
Рис.31. Кинетические кривые осветления суспензии каолина
при введении 0,01 (кривая 1), 0,03 (кривая 2)
и 0,05 масс. % сополимера АА: МАГ (70:30).
Из рис. 31 видно, что резкое снижение мутности суспензии каолина проходит в течение 100-150 с. Этот период времени соответствует осаждению основного количества сформированных в ходе предварительного перемешивания флоккул. Далее оптическая плотность надосадочной жидкости снижается с меньшей скоростью. После осаждения в течение 500 – 600 с остаточная мутность не изменяется.
Начальные скорости осветления суспензии каолина закономерно повышаются при увеличении концентрации полиэлектролита. Скорость осветления в присутствии полимерных добавок выше в 3 – 4 раза, чем скорость осветления в отсутствие полимеров. Наибольшие значения скорости достигаются при дозах 0,05-0,10 мг/г.
Полученные результаты по осаждению 0,5 % суспензии каолина недостаточны для анализа механизма снижения устойчивости при введении исследуемых сополимеров. Представлялось необходимым изучить процессы осаждения при более высокой концентрации дисперсной фазы (0,8%). Повышенное содержание дисперсной фазы позволяет не только оценивать скорость осаждения флоккул, но и определять динамические параметры образующегося осадка. В таблице 23 представлены зависимости объема осадка от времени в присутствии сополимера ААм: МАГ состава 70:30.
Таблица 23
| Образец | Время осаждения, мин. | Объем осадка, мм3 | Остаточная мутность, % |
| 1:99 | 35 | 3,4 | 63 |
| 5:95 | 60 | 4,0 | 56 |
| 70:30 | 15 | 4,5 | 34 |
| Дист. вода+ каолин | 140 | 2,0 | 55 |
Скорость осаждения (накопления осадка) и уплотнения осадка закономерно увеличиваются с повышением количества введенного сополимера.
Сравнивая параметры кинетической устойчивости сополимеров АА с МАГ и ПАА, можно видеть, что сополимеры проявляют значительно большую активность, судя по значениям объема и времени уплотнения осадка. Исходя из известных закономерностей динамики дисперсий, можно предположить, что под действием сополимеров образуются флоккулы большего размера или большей плотности, чем в присутствии полиакриламида.
Известно, что эффективность процесса флоккуляции высокомолекулярными соединениями повышается при добавлении в систему низкомолекулярных электролитов [209]. Низкомолекулярные электролиты, сжимая слой противоионов у поверхности коллоидных частиц и нейтрализуя заряд на их поверхности, облегчают подход макромолекул и их адсорбцию. Одновременно происходит изменение структуры макромолекул. Экранирование заряженных звеньев полимера и уменьшение сил внутримолекулярного отталкивания приводит к сжатию макромолекул. Сжатые макромолекулы, занимая меньший объем, плотнее укладываются на поверхности частиц, в результате чего общее количество адсорбированного полимера возрастает.
В качестве коагулянта использовали органомодифицированную бентонитовую глину месторождения Герпегеж. Выполненные нами опыты показали, что при добавлении флоккулянтов к суспензии каолина, к которой предварительно был добавлен органомодифицированный монтмориллонит (ОМ) резко уменьшалась оптическая плотность раствора, происходило образование и быстрое оседание агрегатов частиц. Этот процесс усиливался с повышением количества добавленного ОМ. Эффективность флоккуляции также зависела от того, в какой последовательности дозируются реагенты – коагулирующий электролит и сополимер. Выявлено, что предварительное введение коагулирующего реагента вызывает более эффективную флоккуляцию (таблица 24).
Таким образом, предварительная агрегация коллоидных частиц позволяет получать крупные хлопья с повышенным содержанием твердой фазы. Однако, хотя процесс флоккуляции протекает достаточно быстро, остаточное значение мутности немного выше при добавлении коагулянта-органоглины.
Таблица 24
Влияние порядка дозирования коагулирующего реагента и сополимеров на эффективность флоккуляции
| Коагулянт | Порядок дозирования | Концентрация сополимера | Остаточная мутность, % |
| ОМ | ОМ+сополимер 70:30 | 0,5 % | 10 % |
| ОМ | сополимер 70:30+ОМ | 0,5 % | 28 % |

а)

б)
Рис. 32: а- осадок суспензии каолина, б-осадок после обработки сополимером МАГ: АА (70:30)
3.6 Определение остаточного полимера в очищенной воде
Для очистки воды могут применяться полимеры, не действующие на человека, животных, фауну и флору водоемов, нетоксичные и малотоксичные.
Существенное влияние на токсичность оказывает количество непрореагировавшего мономера и реагентов используемых при синтезе. Токсичность этих веществ значительно превышает токсичность полимеров. Увеличение молекулярной массы и разветвленность полимера, затрудняющие его диффузию, приводят, по некоторым данным, к снижению токсичности.
Полиакриламид практически нетоксичное вещество [209], а акриламид сильнотоксичное вещество, действующее на центральную нервную систему и ткани дыхательных путей. ПДК для акриламида составляет 156 – 280 мг/кг.
В связи с этим перед использованием для очистки воды высокомолекулярные флоккулянты следует тщательно очищать от низкомолекулярных фракций. В данной работе полученные полимеры многократно переосаждали из воды в ацетон и очищали методом диализа.
При правильно подобранной дозе очищенного от низкомолекулярных веществ флоккулянта и условиях смешения в воде остаются только следы сополимера, который обладает низкой токсичностью по данным исследований с использованием биотестирования на личинках хирономид.
Для определения остаточного сополимера в очищенной воде использовали метод Буркета [210]. Метод основан на добавлении в исследуемую воду суспензии каолина; такое же количество добавляют в стандартные растворы с известным содержанием сополимера. Сопоставляя скорость осаждения или остаточное количество глины в осветленной воде, определяют количество находящегося в ней полимера. Метод позволяет устанавливать содержание высокомолекулярных флоккулянтов до 0,001 – 0,002 мг/л.
Данные, полученные этим методом, показали отсутствие в очищенной воде полимера, что свидетельствует о том, что в исследованных условиях сополимеры практически полностью взаимодействуют с коллоидными частицами. Использование исследованных реагентов для очистки воды, особенно в хозяйственно-питьевом водоснабжении, требует более углубленного изучения токсичности, а также тщательного контроля содержания мономеров.
Таким образом, сочетание в полученных сополимерах высокой бактерицидной активности (за счет содержания гуанидиновых групп) и флоккулирующих свойств позволило нам выявить новые эффективные гуанидинсодержащие биоцидные флоккулянты для очистки и обеззараживания воды.
Литература
1. Кабанов В.А., Топчиев Д.А. // Полимеризация ионизующихся мономеров. – М.: Наука, 1975.
2. Топчиев Д.А., Малкандуев Ю.А. // Радикальная полимеризация N,N – диалкилдиаллиламмоний галогенидов. – Нальчик: КБГУ, 1997.
3. Топчиев Д.А. // Дис. д-ра хим. Наук. – М.: ИНХС, 1973.
4. Кабанов В.А., Зубов В.П., Семчиков Ю.Д. Комплексно-радикальная полимеризация. – М.: Наука, 1987.
5. Butler G.B. // J. Polym. Sci. – 1960. – V.48. – № 1. – P.279.
6. Burtnett M.D., Butler G.B. // J. Org. Chem. – 1960. – V.25. – P.309.
7. Butler G.B. Cyclopolymerization and Cyclocopolymerization. – New York: Marsel Dekker, 1992.
8. Corfield G.C. // Chem. Soc. Rev. – 1972. – V.1. – № 3. – p.523.
9. Butler G.B. //Amer. Chem. Soc. Div., Polym. Chem. Preprints. – 1967. – V.8. – №1. – P.35.
10.Butler G.B., Kimura S. // J. Macromol. Sci. Chem. A. – 1971. – V.5. – №1. – p.181.
11.Butler G.B., Crawshow A., Miller W.L. // J. Am. Chem. Soc. – 1958. – V.80. – №14. – p.3165.
12.Julia M., Maumy M. // Bull. Soc. Chim. Fr. 1966. – V.1. – p.434.
13.Julia M. // Chem. Eng. News. – 1966. – V.41. – P.100.
14.Butler G.B. // J. Am. Chem. Soc. – 1967. – V.89. – P.35.
15.Richey H.G., Rothman A.M. // Tetrahedron Lett.. – 1968. – V.12. – P.1457.
16.Brace N.O. // J. Polym. Sci.. – A-1. – 1970. – V.8. – № 8. – P.2091.
17.Lancaster J.E., Baccei L., Panzer H.P. // J. Polym. Sci., Polym. Lett. Ed. – 1976. – V.14. – № 9. – P. 549.
18.Gibbs W.E., Barton J.M. / in book: Kinetics and Mechanism of Polymerization/ Ed. Hat G.E. – New York: Dekker, 1978, part 1, chapter 2.
19.Panzik H.L., Mulvaney J.E // J. Polym. Sci., Polym. Chem. Ed. –1972. – V.10. – № 12. – P.3469.
20.Uzushido K., Matsumoto A., Giwa M. // J. Polym. Sci., Polym. Chem. Ed. – 1978. – V.16. – № 5. – P.1081.
21.Gray T.F., Butler G.B. // J. Macromol. Sci. Chem. A. – 1975. – V9. – № 1. – P.45.
22.Matsumoto A., Tamura J., Jamawak M., Oiwa M. // J. Polym. Sci., Polym. Chem. Ed. –. 1979. – V.17. – № 5. – P.1419.
23.Johns S.R., Willing R.I., Middleton S., Ong A.K. // J. Macromol. Sci. Chem. A, 1979, V.10, № 5, p.875.
24.Ottenbreit R.M. // Ing. Engng. Chem. Prod. Res. Dev. 1980, V.19, p.520.
25.Bouman L.M., Cha C.I. // J. Polym. Sci. Polym. Lett. Ed. 1979, V.17, № 3, p.167.
26.Wandrey C. // Acta Polym. 1981, V.32, p. 177.
27.ТопчиевД.А., НажметдиноваГ.Т., КрапивинА.М., ШрейдерВ.А., КабановВ.А. // Высокомолек. соед. Б, 1982, Т.24Б № 6, с. 473.
28.Solomon D.H. // J. Macromol. Sci. Chem. A, 1975, V.9, № 1, p.97.
29.Hawthorne D.G., Johns S.R., Willing R.I. // Aust. J. Chem. 1976, V.29, № 9, p.315.
30.Johns S.R., Willing R.I., Middleton S., Ong A.K. // J. Macromol. Sci. Chem. A, 1979, V.10, № 5, p.875.
31.Hawthorne D.G., Johns S.R., Solomon D.H. Willing R.I. // Aust. J. Chem. 1979, V.3, № 215, p.1155.
32.Beckwith A.L., Ong A.K., Solomon D.H. // J. Macromol. Sci. Chem. A,. 1975, № 9, p.125.
33.Beckwith A.L., Hawthorne D.G., Solomon D.H. // Aust. J. Chem. 1976, V.29, № 9, p.995.
34.Solomon D.H. // J. Polym. Sci. Polym. Symposium. 1975, V.49, p.175.
35.Haman S.D., Pompe A., Solomon D.H., Spurling T.H. // Aust. J. Chem. 1976, V.29, № 9, p.1975.
36.Moad G., Solomon D.H. // Chemistry of free radical polymerization. Oxford: Pergamon, 1995.
37.ТопчиевД.А., БикашеваГ.Т., МартыненкоА.И., КапцовН.М., ГудковаЛ.А., КабановВ.А. // Высокомолек. соед. Б. 1980, Т.22, № 4, с.269.
38.Топчиев Д.А., Бикашева Г.Т., Мартыненко А.И., Капцов Н.М., Гудкова Л.А., Кабанов В.А. Полимерные амины: синтез мономеров, полимери-зация и пути использования в народном хозяйстве. М.: Наука, 1980.
39.Нажметдинова Г.Т. Дис. канд. хим. наук. М.: ИНХС, 1983.
40.Нажметдинова Г.Т., Шрейдер В.А., Топчиев Д.А., Кабанов В.А. // Изв. АН СССР. Сер.хим. 1984, Т.5, с.1024.
41.Топчиев Д.А., Нажметдинова Г.Т. // Высокомол. соед. А, 1983, Т. 25, №3, с.636.
42.Топчиев Д.А., Нажметдинова Г.Т., Кабанов В.А. // Изв. АН СССР. Сер. хим. 1989, Т.9, с.2146.
43.Babaev N.A., Martynenko A.I., Topchiev D.A., Kabanov V.A., Wandrey Ch., Hahn M., Jaeger W., Reinisch G. // Acta Polymerica. 1985, V.36, № 7, p.396.
44.ГолубковаН.А., МартыненкоА.И., БабаевН.А., НечаеваА.В., ЭфендиевА.А., ТопчиевД.А., КабановВ.А. // Изв. АНСССР, сер.хим., 1986, Т. 2, с.485.
45.Малкандуев Ю.А., Коршак Ю.В., Микитаев А.К, Топчиев Д.А., Кабанов В.А. // Материалы V Международного микросимпозиума «Радикальная полимеризация». Уфа, 1984, с.46.
46.Бабаев Н.А., Мартыненко А.И., Оппенгейм В.Д., Крапивин A.M., Эфендиев А.А., Топчиев Д.А. // Азерб. хим. журн. 1983, Т.4, с.89.
47.Мартыненко А.И., Вандрей К., Егер В., Хан М., Топчиев Д.А., Райниш Г., Кабанов В.А. // Материалы. V Международного микросимпозиума «Радикальная полимеризация». Уфа, 1984, с.74.
48. Четыркина Г.М., Соколова T.A., Котон М.М. // ВМС. I960. Т. 2. № 8. С. 1207-1212.
49. McCormiek C.L., Johnson СВ. //Polymer Mater. Sci. Eng. 1986. V.55. P.366-370; Chem. Abstr. 1986. V. 105.191686.
50. Morishima Y., Itoh Y., Nozakura S. //Makromol. Chem. 1981. Bd. 182. N11. S.3135-3147.
51. Iton Y., Morishima Y„ Nozakura S. //J. Polymer Sci. Polym. Chem. Ed. 1982. V. 20. N2. P. 467-476.
52. Kurenkov V.F., Myagchenkov V.A. //Acta Polymeiica. 1986. Bd. 37. N8, S. 517-524.
53. Ilavsky M. // Macromolecules. 1982. V. IS. N3. P. 782-788.
54. Стародубцев СП, Рябина B.P. //ВМС. Сер. А. 1987. Т. 29. № 11. С. 2281-2285.
55. Salamone J.C., Tsai С.С., Watterson А.С. et al. Polymeric Amines and Ammonium Salts/Ed. by E.J.Goethals. Oxford: Pergamon Press, 1980. P. 105-112.
56. Salamone J.C., Mahmud N.A., Mahmud M.V. et al. //Polymer. 1982. V. 23. N6. P. 843-848.
57. Bock J., Siano D.B., SchuUD.Het al. //J. Polymer Mater. Sci. Eng. 1986. V. 55. P. 355-360; Chem. Abstr. 1986. V. 105.191685.
58. McCormiek C.L., Nonaka Т., Johnson СВ. // Polymer. 1988. V. 29. N 4. P. 731-739.
59. McCormiek C.L., Hutchinson B.H., Morgan S.E. //Makromol. Chem. 1987. Bd. 188. N 2. S. 357-370.
60. Буянов АЛ., Рееелъская Я.Г., Петропавловский Г.А. и др. // ВМС. Сер. Б. 1989. Т. 31. № 12. С. 883-887.
61. Nyquist В.Е. Functional Monomers. Their Preparation, Polymerization and Application /Ed. by R.H.Yokum. – New York: M. Decker, 1973. – V. 1.– 745p.
62. Kamogawa H., Sekiyo T. I. // Polymer Sci. 1961. V.50. N 153. P. 211-225.
63. Глазомицкий КМ., Кирпиченко K.P., Гольцин Б.Э. и др. //ЖПХ. 1971. Т. 44. № 5. С. 1192-1195.
64. Feldman D., Unguretne С, Crusos A. et al. //Mater, plast. 1976. Т. 13. N 2. P. 69-73; РЖХ. 1977.4T739.
65. Bonardi С, Boutevin В., Pietrasanta Y, et al. //Makromol. Chem. 1985. Bd. 186. N2. S. 261-271.
66. Бектуров E.A., Мягченкое B.A., Куренков В.Ф. Полимеры и сополимеры стирол-сульфокислоты. – Алма-Ата: Наука, 1989. – 200 с.
67. Мягченков B.A., Френкель С.Я. Композиционная неоднородность сополимеров. – Л.; Химия, 1988. – 248 с.
68. Daintan F.S., Sisley W.D. //Tram. Faraday Soc. 1969. V. 59. N 486. Part 6. P. 1385-1389.
69. Hart R., Ttmmerman D. //J. Polymer Sci. I960. V. 48. N 150. P. 151-157.
70. Otsu Т., Inove M., Yamada B. et al. // J. Polymer Sci. Polymer Lett. Bd. 1975. V. 13. N 8. P. 505-510.
71. Петровa Г.А. ., ШтрайхманГ.А., ВаншейдтА.А. //ЖФХ. 1959. Т. 33. № 6. С. 1246-1252.
72. Yokoda К., Oda J. //Kogyo Kagaku Zasshi. – 1970. – V.73. – N1. – P. 224-228; Chem. Abstr. – 1970. – V. 72.– 122003.
73. Whistler R.L, GoatleyJ. // J. Polymer Sci. 1961. V. 50. N153. P. 127-132.
74. Sur G.S.,Noh S.K., Choi S.K. //. Polymer Sci., Polymer Chem. Ed. 1981. V. 19. N 2. P. 223-233.
75. Wichterle O., Gregor V. //J. Polymer Sci. 1959. V. 34. N127. P. 309-317.
76. Bork J.F., Wyman D.P. et al. //J. Appl. Polymer. Sci. – l963. – V.7. – N2. – P.451-459.
77. Jordan E.F., Bennett R„ Shumon A.C. et al. // J. Polymer Sci. A-l. 1970. V. 8. N 11. P. 3113-3121.
78. Oishi Т., FujimotoM.Hl. //Polymer Sci. Polymer Chem. Bd. 1982. V.20. N9. P. 2727-2730.
79. Yomada В., Yoshiokt M., Otsu T. //J. Polymer Sci. Polymer Chem. Ed. 1984. V. 22. N 2. P. 463-473.
80. Соколова T.A., Четыркина Г.М. // BMC. 1961. T. 3. № 2. C. 244-547.
81. Glpstein В., Hewett W.A., Need O.U. //J. Polymer Sci. A-l. 1970. V. 8.N 11. P. 3285-3294.
82. Zabransky J, Houske M., KalotJ. // Makromol. Chem. 1985. Bd. 186. N2. S. 247-253.
83. Наволокина P.А., Зилъберман E.H., Кузнецова Т.А. //Физико-хим. основы синтеза и переработки полимеров. – Горький: Изд-во ГГУ, 1983. – С. 15-17.
84. Watanabe К, Sekei М„ SakakibaraY. etal. //Kogyo Kagaku Zasshi. 1970. V. 73. N 5. P. 1056-1058; Chem. Abstr. 1970. V. 72.122003.
85. Xван P.M., Мусаев У.Н., Бабаев T.M. и др. //Физико-хим. основы синтеза и переработки полимеров. Горький: изд-во ГГУ, 1979. С. 13-18.
86. Мягчевкое В.А., Френкель С.Я. //Успехи химии. 1973. Т. 42. № 5. С.827—853.
87. Мягченков В.А., Френкель С.Я. //Усп. химии. 1978. Т. 47. N* 7. С. 1261-1292.
88. Saint G„ Leoni A., Franco S.Y. //Маkromol. Chem. 1971. Bd. 144. S. 235-244.
89. Chung K.H. Hi. //Korean Chem. Soc. 1970. V. 14. N 4. P. 333-339; Chem. Abstr, 1971. V. 75.118647.
90. Winsfc L.M., Kotlarchik C, Darlak R.S. // J. Polymer Sci. Polymer Chem. Ed. 1973. V. 11. N 2. P. 353-365.
91. Minsk K.M., Kotlarchik C, Meyer G.N. // J. Polymer Sci. Polymer Chem. Ed. 1973. V. 11. N12. P. 3037.
92. Jacob M., SmetsG., DeSchryverF. // J. Polymer Sci. B. 1972. V. 10. N 9. P. 669-673.
93. Chatterjee A.M., Burns CM. //Can. I. Chem. 1971. V.49. N 20. P. 3249-3251.
94. Kamogawa H. //Kogyo Kagaku Zasshi. 19S8. V. 61. P. 1024-1027; Chem. Abstr. 1961. V. 55. P. 17077.
95. Saini G., Leoni A., Franco S. //Makromol. Chem. 1971. Bd. 147. S. 213-218.
96. Leoni A., Francos., Saint G. //Makromol. Chem. 1973. Bd. 167. S. 97-104.
97. Franco S., Leoni A. //Polymer. 1973. V. 14. N1. P. 2-4.
98. Knobloch F.W. //J. Polymer Sci. 1957. V. 25. N10. P. 453-464. 297.
99. Soini G., Leoni A., Franco S. //Makromol. Chem. 1971. Bd. 146. S. 165-171.
100. Kurenkov V.F., Myagchenkov V.A. //Europ. Polym. J. 1980. V. 16. N 11. P. 1229-1239.
101. Хам Д. Сополимеризация. – M.: Химия, 1971.616 с.
102. North A.M., Scallam A.M. //Polymer. 1964. V.5. N 9. P. 447-455.
103. Saini G., Polla-Muttiot G., MeironeM. //J. Polymer Sd. 1961. V. 50. N 153. P. 812-813.
104. АбрамоваЛ.И., Зилъберман E.H., ЧугуноваЛ.С. //ВМС. Б. 1979. Т. 21. № 11. С. 813-817.
105. Fineman М., RossS.D. H. //J. Polymer Sci. 1950. V. 5. N 2. P. 259-262.
106. Наволокина P.A., Зилъберман Б.Н., Масленникова Т.И. //ВМС. Сер. Б. 1982. Т. 24. Я* 8. С. 609-612.
107. Зилъберман Е.Н., Наволокина Р.А., Абрамова Н.А. //ВМС. Сер. Б. 1983. Т. 25. № 4. С. 279—283.
108. Kelen Г., Tudos Р„ Turmnyi В. et at.//J. Polymer Sci. Polymer Chem. Ed. 1977. V. 15. N12. P. 3047-3074.
109. Tidwell P.W., Mortimer СA. //J… Polymer Sci. A. 1965. V. 3. N1. P. 369-387.
110. Park K. Y.r Santee E.R., Harwood H.J. //Polymer Prepr. 1986. V. 27. N 2. P. 81-82.
111. Зилъберман Б.Н., Абрамова Л.И., Черненкова ЮМ. и др. //ВМС. Сер. А 1984. Т. 26. № 7. С. 1365-1368.
112. Оудиан Дж. Основы химии полимеров. М.: Мир, 1974, 614 с.
113. Hirooka М., Yabuuchi Я., Kawasumi S, et al. //J. Polymer Sci. Polymer Chem. Ed. 1973. V. 11. N 6. P. 1281-1306.
114. Мягченков B.A., ФренкельС.Я. //Усп. химии. 1968. Т. 37. №» 12. С. 2247-2271.
115. Куренков В.Ф., Верижникова А.С., Кузнецов Е.В. и др. //Химия и хим. технология. 1982. Т. 25. № 2. С. 221-226. (Изв. вузов).
116. Мягченков В.А., Ларионова Л.А., Вагапова А.К. и др. //ДАН СССР 1972. Т. 207. С. 337-380.
117. Басова Т.Г., Зилъберман Е.Н., Шварева Г.Н. и др. //ВМС. Сер. Б. 1975. Т. 17. № 5. С. 379-383.
118. Мягченкое В.А., Куренков В.Ф., Френкель С.Я. //ВМС Сер. А. 1968. Т. 10. IP 8. С. 1740-1748.
119. Мягченкое В.А., Куренков В.Ф., Кузнецов Б.В. и др. //ВМС. Сер. А. 1969. Т. 11. К" 8. С. 1789-1792.
120. Мягченкое В.А., Куренков В.Ф., Френкель С.Я. //ДАН СССР 1969. Т. 184. №> 4. С. 880-882.
121. Мягченкое В.Л., Вагапова А.К., Кузнецов Е.Л. и др. //ДАН. 1973. Т. 210. № 1. С. 151-153.
122. Куренков В.Ф. //Химия и технол. элементоорганич. соед. и полимеров. 1974. Т. 314. С. 113-117.
123. Kurenkot V.F., Akhmedjanova R.A., Myagchenkov V.A. // Aсtа Polymerica. 1981. Bd. 32.N 10. S. 612-615.
124. Мягченков В.А., Куренков В.Ф., Кукушкина И.А. и дp. //ДАН СССР. 1977. Т. 236. № 1. С. 157-160.
125. Куренков В. Ф., Кукушкина И.А., Кузнецов Е.В. и др. // Химия и технол. элементо-органич. соед. и полимеров. Т. 5. Казань: КХТИ, 1976, С. 72-76.
126. Myagchenkov V.A., Vagapova А.К., Kurenkov V.F. et al. //Europ. Polymer. J. 1978. V. 14. N 2. P. 169-171.
127. Myagchenkov V.A., Kurenkov V.F., Frenkel S.Ya. //Acta Polym. 1982. Bd. 33. N6. S. 388-394.
128. Kurenkov V.F., Myagchenkov V.A. //Acta Polym. 1986, Bd. 37. N 7. S. 424-435.
129. Rajan C.R., Srinivas Y.S., Ponrathman S. et al. //J. Polymer Sci. Polymer Lett. 1987. V. 25. N 2. P. 73-77.
130. КуренковВ.Ф., Кузнецов E.B,, Мягченков B.A. // Тр. Казанскогохим.-технол. ин-та. Казань: КХТИ, 1969. № 40. Часть II. С. 38-43.
131. Truong N.D., Calin J.C., Francois J. et al. //Polymer. Y. 1986. V. 27. N3. P. 467-475.
132. Басова Т.Г., Зилъберман E.H., Шварева ГЛ. и др. // ВМС. Сер. Б. 1977. Т. 19. №1. С. 22-25.
133. Кабанов В. А., Топчиев Д. А. Полимеризация ионизующихся мономеров. – М.: Наука, 1975. – 224 с.
134. Barton I, Gutanicova V. //Chem. Papers, 1987. V.41. N1. P. 113-117.
135. Зилъберман E.H., Новолокина P.A., КубарэтаО.П. //ВМС. – 1980. – Т.22. – №9 – С. 2006-2011.
136. Каргин В.А., Платэ H.A… Патрикеева Т.П. //ВМС. 1964. Т. 6. № 11. С. 2040-2045.
137. Abe Z., Unaka Н., Sumimoto М. //J. Polymer Sci. Polymer Chem. Ed. 1978. V. 16. N1. P.305-308.
138. Танака H. //J. Polym. Sci. Polym. Chem. Ed. 1986. V. 24. N1. P. 29-36.
139. Yukawa J., Kataoka K., Tsuruta T. //Polymer J. 1979. V. 11. N11. P. 895-900.
140. Shyluk W.P. //J. Polymer Sci. A. 1964. V. 2. N 5. P. 2191-2206.
141. Зильбермон E.H., Черненкова Ю.И., Шварева Г.Н. и др. // Физ.-хим. основы синтеза и переработки полимеров. № 3. Горький: изд-во ГТУ, 1978. С. 20-22.
142. Черненкова Ю.П., Зилъберман Е.Н., Шварева Г.Н. // Физ-хим. основы синтеза и переработки полимеров. Горький: иэд-во ГГУ, 1984. С. 27-31.
143. Черненкова Ю.П. Синтез сополимеров на основе диалкиламиноалкилметакрила-тов: Автореф. Дис.… канд. хим. наук. Казань: Казанский хим.-технол. ин-т, 1984. 32 с.
144. Черненкова Ю.П., Зилъберман Е.Н., Шварева Г.Н. //ВМС. Сер. Б. 1982. Т. 24. № 2.ь С. 119-122.
145. Черненкова Ю.П., Зилъберман Е.Н., Шварева Г.Н. и др. //НЖПХ. 1980. Т. 53. № 2.J С. 378-382.
146.Яминский В.В. и др. Коагуляционные контакты в дисперсных системах. – М.: Химия, 1982.
147. Фридрихсберг Д.А. Курс коллоидной химии. – М.: Химия, 1974.
148. Небера В.П. Флоккуляция минеральных суспензий. – М.: Недра, 1983.
149. Левич В.Г. Физико-химическая гидродинамика. – М: Физмат, 1959.
150. Баран А.А., Соломенцева И.М. Флоккуляция дисперсных систем водорастворимыми полимерами и ее применение в водоочистке. //Химия и технология воды. – 1983. – т.5. – 32.
151. Бектуров Е.А., Бимендина Л.А. Интерполимерные комплексы. – Алма-Ата: Наука, 1977.
152. Вейцер Ю.И. и др. Использование катионных полиэлектролитов для очистки воды от вирусов. //Гигиена и санитария. – 1976. – С. 3.
153. Эпштейн С.И., Пантелят Г.С. К вопросу разрушения хлопьев взвешенных веществ в турбулентном потоке. – В кн.: Вопросы технологии обработки воды промышленного и питьевого водоснабжения. – Киев: Будивельник, 1969.
154. Полиакриламид / Под ред. В.Ф.Куренкова. – М.: Химия, 1992. 192 с.
155. Небера В.П. Флоккуляция минеральных суспензий. – М.: Недра, 1983. 288 с.
156. Вейцер Ю.И., Минц Д.М. Высокомолекулярные флоккулянты в процессах очистки природных и сточных вод. – М.: Стройиздат, 1984. с. 202.
157. Баран А.А. Полимерсодержащие дисперсные системы. – Киев: Наук. думка, 1986. с. 204.
158. Николаев А.Ф., Охрименко Г.И. Водорастворимые полимеры. – Л.: Химия, 1979. с. 61.
159. Halverson F., Panzer H.P. // Kirk-Othmer Encyclopedia of Chemical Technology. 3rd ed. – N.Y.: Wiley, 1980. – Vol. 10. – P. 489.
160. Полиакриламид / Подред. В.Ф. Куренкова. – М.: Химия, 1992. 192 с.
161. Kurenkov V.F., Myagchenkov V.A. // Polymeric Materials Encyclopedia. Boca Raton (Fla): CRC Press Inc., 1996. Vol. 1.
162. Alfrey T., Overberger C.G., Pinner S.H. // J. Am. Chem. Soc. 1953, V.75. – p.4321.
163. СивовН.А., МартыненкоА.И., КабановаЕ.Ю., ПоповаН.И., ХашироваС.Ю., ЭсмурзиевА.М. // Нефтехимия. – 2004. – №1. – C. 47-51.
164. ХашироваС.Ю., СивовН.А., ПоповаН.И., КабановаЕ.Ю., МартыненкоА.И., МалкандуевЮ.А., ТопчиевД.А. Синтезновыхмономеров на основе диаллилгуанидина и их способность к радикальной полимеризации. // Известия вузов. Сев.-Кавк. Регион. Сер. Естеств. Науки. 2002. № 3. С. 82-85.
165. Малкандуев Ю.А., Хаширова С.Ю., Сивов Н.А., Попова Н.И., Кабанова Е.Ю., Мартыненко А.И., Топчиев Д.А. Некоторые особенности сополимеризации N,N-диаллилгуанидинацетата и N,N-диаллил- N,N-диметиламмонийхлорида. // Известия вузов. Сев.-Кавк. Регион. Сер. Естеств. Науки. 2003. №1. С. 19-22.
166. П.А.Гембицкий, И.И.Воинцева. Полимерный биоцидный препарат. – Запорожье: Полиграф, 1998. – 42 с.
167. Хаширова С.Ю., Эсмурзиев А.М., Мартыненко А.И., Кабанова Е.Ю., Попова Н.И., Сивов Н.А., Малкандуев Ю.А. Радикальная гомо(со)полимеризация акрилат- и метакрилатгуанидинов в водных средах //Известия вузов. Сев.-Кавк. Регион. Сер. Естеств. науки. 2004. №1. С. 82-85.
168. Бьерклунд Ю.А., Рейтерхэлл А.Р. Патент № 417569, Швеция, 1981.
169. Химическая энциклопедия / под ред. И.Л.Кнунянца. 1988, M., Т. 2 С. 138.
170. Платэ Н.А., Васильев А.Е. Физиологически активные полимеры. – М.: Химия, 1986, – c. 296.
171. Платэ Н.А., Васильев А.Е. // Высокомолек. соед. А, 1982, Т.24, №4, с.675.
172. Ryser H.J. // Science. 1965, V.150, p.501.
173. Ryser H.J. // Biomembranes. 1971, V.2, p.197.
174. Ярославов А.А., Кабанов В.А. // Материалы Всероссийского Каргинского симпозиума. 2000. Тез. докл. ч.1, с.17.
175. Фельдштейн М.М. // Синтетические полимеры медицинского назначения. Материалы 6 Всесоюзного симпозиума. Алма-Ата, 1983, с.142.
176. Сивов Н.А., Мартыненко А.И., Кабанова Е.Ю., Попова Н.И., Хаширова С.Ю., Эсмурзиев А.М. // Нефтехимия, №1, 2004, с. 47-51.
177. Хаширова С.Ю., Сивов Н.А., Попова Н.И., Кабанова Е.Ю., Мартыненко А.И., Малкандуев Ю.А., Топчиев Д.А. Синтез новых мономеров на основе диаллилгуанидина и их способность к радикальной полимеризации. // Известия вузов. Сев.-Кавк. Регион. Сер. Естеств. Науки. 2002. № 3. С. 82-85.
178. Малкандуев Ю.А., Хаширова С.Ю., Сивов Н.А., Попова Н.И., Кабанова Е.Ю., Мартыненко А.И., Топчиев Д.А. Некоторые особенности сополимеризации N,N-диаллилгуанидинацетата и N,N-диаллил- N,N-диметиламмонийхлорида. // Известия вузов. Сев.-Кавк. Регион. Сер. Естеств. Науки. 2003. №1. С. 19-22.
179. П.А.Гембицкий, И.И.Воинцева. Полимерный биоцидный препарат. – Запорожье: Полиграф, 1998. – с. 42 .
180. Хаширова С.Ю., Эсмурзиев А.М., Мартыненко А.И., Кабанова Е.Ю., Попова Н.И., Сивов Н.А., Малкандуев Ю.А. Радикальная гомо(со)полимеризация акрилат- и метакрилатгуанидинов в водных средах //Известия вузов. Сев.-Кавк. Регион. Сер. Естеств. науки. 2004. №1. С. 82-85.
181. Бьерклунд Ю.А., Рейтерхэлл А.Р. Патент № 417569, Швеция, 1981.
Химическая энциклопедия / под ред. И.Л.Кнунянца. 1988, M., Т. 2 С. 138.
182. Сополимеризация / под ред. Д.Хэма, 1971, М., Химия, с. 12.
183. Alfrey T. Goldfinger G. // J. Chem.Phys. 1944. V.12, N 6. р.205
184. Езриелев А.И., Брохина Э.Л., Роскин Б.С. // Высокомол. соединения 1969, А, Т.11, №8. с.1670.
185. Fineman M., Ross S.D. // J. Pol. Sci. 1950. V.5. р.251
186. Khokhlov A.R., Khalatur P.G. // Physica A. 1998, V.249, p.253.
187. Khokhlov A.R., Khalatur P.G. // Phys. Rev. Lett. 1999, V.82, p.3456.
188. ЛозинскийВ.И., СименелИ.А., КурскаяЕ.А., КулаковаВ.К., ГринбергВ.Я., ДубовикА.С., ГалаевИ.Ю., МаттиассонБ., ХохловА.Р. // ДАН, 2000, т.375, №5, с. 637.
189. Г.Е.Афиногенов, Е.Ф.Панарин // Антимикробные полимеры. – СПб: Гиппократ, 1993, с. 261.
196. Брагинский Л.П. Методологические аспекты токсикологического биотестирования на Daphnia magna Str. и других ветвистоусых ракообразных (критический обзор) // Гидробиол. журн. – 2000. – Т. 36, № 5. – С.50-70.
197.Смирнов Н.Н. Биология ветвистоусых ракообразных / Зоология беспозвоночных – 3. – М., 1974. – 115 с. (Итоги науки и техники, ВИНИТИ АН СССР).
198.Метелев В.В., Канаев А.И., Дзасохова Н.Г. Водная токсикология. – М.: Колос, 1971. – 246с.
199. Вейцер Ю.И., Минц Д.М. Высокомолекулярные флоккулянты в процессах очистки природных и сточных вод. – М.: Стройиздат. 1984. 202с.
200. Баран А.А., Соломенцева И.М. Флоккуляция дисперсных систем водорастворимыми полимерами и ее применение в водоочистке. //Химия и технология воды. – 1983. – т.5. – C. 32.
201. Булидорова Г.В., Мягченков В.А. Кинетические особенности седиментации каолина в присутствии анионного и катионного полиакриламидного флоккулянтов // Коллоид, журн. — 1995. — Т.57, N6. -С. 778-782.
202. Булидорова Г.В., Мягченков В.А. Кинетика седиментации каолина при совместном введении флоккулянта (катионного полиакриламида) и коагулянта // Коллоид, журн. — 1996. — Т.58, N1. — С.29-34.
203. Gregory J. Turbidity fluctuations in flowing suspentions // J. Colloid and Interface Sci. — 1985. — V.105, N2 — p. 357.
204. Барань Ш. (Баран A.A.), Грегори Д. Флоккуляция суспензии каолина катионными полиэлектролитами // Коллоид. журн. — 1996. -Т.58, N1.-С. 13-18.
205. Куренков В.Ф., Чуриков Ф.И., Снегирев СВ. Седиментация суспензии каолина в присутствии частично гидролизованного полиакриламида и А12(804)з. // ЖПХ, 1999 — т. 72, № 5. — С. 828-833.
206. Куренков В.Ф., Шарапова З.Ф., Хайрулин М.Р и др. Влияние молекулярных характеристик натриевой соли 2-акриламидо-2-метилпропан сульфокислоты с N-винилпирролидоном на флоккулирующие свойства. // ЖПХ. – 1999. — т. 72, № 8. — с. 1374-1379.
207. Куренков В.Ф., Снегирев СВ., Древоедова Е.А, Чуриков Ф.И. Исследование флоккулирующих свойств полиакриламидных флоккулянтов марки Praestol. // ЖПХ. – 1999. — Т. 72. – № 11. — С. 1892-1899.
208. Мягченков В.А, Баран А.А, Бектуров Е.А. и др. Полиакриламидные флоккулянты. – Казань.: Казан, гос. тех. ун-т.-1998,-288 с.
209. Burcket H. Die Bestimung von Spuren Polyacrylamid in Wasser, Gas und Wasserflach, 1970, III, 5.